Article Text

Abstract
Introduction Amyotrophic lateral sclerosis (ALS) is an adult-onset, progressive and universally fatal neurodegenerative disorder. In Europe, Australia and Canada, riluzole is the only approved therapeutic agent for the treatment of ALS, while in the USA, riluzole and edaravone have been approved by the Food and Drug Administration (FDA) . Neither riluzole nor edaravone treatment has resulted in substantial disease-modifying effects. There is, therefore, an urgent need for drugs that result in safe and effective treatment. Here, we present the design and rationale for the phase 2 RESCUE-ALS study, investigating the novel nanocatalytic drug, CNM-Au8, as a therapeutic intervention that enhances the metabolic and energetic capacity of motor neurones. CNM-Au8 is an aqueous suspension of clean-surfaced, faceted gold nanocrystals that have extraordinary catalytic capabilities, that enhance efficiencies of key metabolic reactions, while simultaneously reducing levels of reactive oxygen species. This trial utilises a novel design by employing motor unit number index (MUNIX), measured by electromyography, as a quantitative measure of lower motor neurone loss and as an early marker of ALS disease progression.
Methods and analysis This is a multicentre, randomised, double-blind, parallel group, placebo-controlled study of the efficacy, safety, pharmacokinetics and pharmacodynamics of CNM-Au8 in ALS patients. Patients will be randomised 1:1 to either receive 30 mg of CNM-Au8 once daily or matching placebo over a 36-week double-blind treatment period. Efficacy will be assessed as the change in motor neurone loss as measured by electromyography (eg, MUNIX, the primary endpoint; and secondary endpoints including MScanFit, motor unit size index, Split Hand Index, Neurophysiology Index). Exploratory endpoints include standard clinical and quality of life assessments.
Ethics and dissemination RESCUE-ALS was approved by the Western Sydney Local Health District Human Research Ethics Committee (Ethics Ref: 2019/ETH12107). Results of the study will be submitted for publication in a peer-reviewed journal.
Trial registration number NCT04098406
- motor neurone disease
- clinical trials
- neuromuscular disease
- adult neurology
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
Description of the first oral nanotherapeutic for amyotrophic lateral sclerosis (ALS) patients.
Description of the drug’s mechanism of action, including both a robust activity that supports cellular energy metabolism, as well as nanocatalytic antioxidant activity for neuroprotection.
A novel protocol using electromyography to monitor decreases in functional motor neurone numbers (motor unit number index), an electrophysiological biomarker which predicts ALS disease progression. This design allows for a relatively small sample size to determine rate of change in predicted disease progression.
Aim of the study is to yield results to form the basis and rationale for proceeding to a larger phase III randomised controlled trial.
Introduction
Amyotrophic lateral sclerosis (ALS) is a universally fatal neurodegenerative disorder due to progressive loss of motor neurones in the cerebral cortex, brain stem and spinal cord.1 ALS, also known as Lou Gehrig’s or motor neurone disease, leads to the death of the neurones controlling voluntary muscles, resulting in weakness, muscle atrophy and progressive paralysis. The diagnosis is clinically based and requires the identification of upper and lower motor neurone abnormalities.2 Death typically occurs as early as 3–5 years postdiagnosis.3
In 2015, there were an estimated 222 801 cases of ALS worldwide.4 This has been projected to increase by 69% to 376 674 cases by 2040, in part due to an increasing ageing population. ALS is not only a devastating neurological disease with no cure; the economic burden associated with this disease is considerable, with an estimated annual total cost of $69 475 per affected individual as of 2015 in the USA.5 6
Currently, the two FDA-approved therapeutic agents for the treatment of ALS are riluzole, an antiglutamatergic agent7 and edaravone, a free-radical scavenger.8 However, both of these drugs have demonstrated limited disease-modifying effects. Riluzole extends patient lifespans by an average of 2–3 months, while edaravone slows the rate of decline in the ALS Functional Rating Score-Revised (ALSFRS-R) score in only a small subset of patients treated at an early stage of disease.9 There is clearly an urgent unmet need for the development of safe and effective disease-modifying therapeutics for ALS.
While most ALS cases are sporadic (sALS) in which the cause remains unknown, about 10%–15% of cases are familial forms of the disease (fALS).10 Most familial cases arise from variants in single genes. To date, over 50 such ALS genes or gene candidates have been discovered,11 including SOD1, TDP43, FUS or C9ORF72. For the most part, fALS is inherited in an autosomal-dominant fashion and is clinically indistinguishable from sALS.12 The functions of identified fALS genes encompass RNA processing, metabolism, regulation of protein aggregation, maintenance of mitochondrial function and protection against oxidative stress.11
More recently, research has uncovered functional commonalities among the seemingly disparate pathways of metabolism that are impaired in ALS. Dysregulation of these pathways are indicative of an underlying unifying pathogenetic mechanism of bioenergetic failure in ALS.13 Bioenergetic effects in ALS affect motor neurones and also the astrocytes and oligodendrocytes, which provide essential trophic and nutritional support to motor neurones. Postmortem analyses of brain tissues from both fALS and sALS patients have revealed widespread accumulation of oxidative damage to proteins, lipids and DNA similar to transgenic mice expressing mutant SOD1 forms, including spinal cord tissue damage with excessive protein and lipid oxidation.14 Such indicators of oxidative stress can also be associated with bioenergetic failure.
Ultimately, ALS is a disease of energetic dysmetabolism involving neurones that have become exquisitely sensitive to apoptosis since their energetic demands are not being met.13 15–18 The main energy-producing organelle of the cell, the mitochondrion, is dysfunctional in ALS, and there are fewer mitochondria in the spinal cords of both fALS and sALS patients.19 Given the clear involvement of cellular bioenergetic failure in the pathogenesis of ALS, the development of a disease-modifying therapeutic that directly addresses the energetic dysregulation underlying progressive neurodegeneration is a rational therapeutic strategy.
CNM-Au8 is a concentrated, aqueous suspension of clean-surfaced faceted nanocrystalline gold that has received regulatory approval to proceed into phase 2 clinical studies in patients with multiple sclerosis, ALS and Parkinson’s disease. These clinical trials are currently ongoing, and are registered on clinicaltrials.gov. Investigational New Drug Application (IND)-enabling toxicology studies and phase 1 first-in-human studies demonstrated that CNM-Au8 had no toxicity findings in animals and a very acceptable safety profile in humans. CNM-Au8 has been demonstrated to be an efficient catalyst for metabolic energy reactions, converting the energetic metabolite nicotinamide adenine dinucleotide hydride (NADH) into NAD+ in proof-of-principle cell-free assays.20 This activity resulted in increased intracellular NAD+ and increased ATP production in primary neuronal–glial cocultures in vitro.20 Application of CNM-Au8 to oligodendrocyte precursor cells was sufficient to induce the transcription of a number of genes involved in the proteostatic Heat Shock Factor 1 (HSF1) response.11 The increased availability of NAD+ and ATP, in addition to increased HSF1 activity, have been shown to be neuroprotective in ALS disease models.21–24 Specifically, oral gavage dosing of CNM-Au8 in SOD1 transgenic mice, used to model one of the fALS disease states, resulted in improved clinical scores throughout the lifespan, and extended the median lifespan of treated animals by 23 days when compared with placebo-treated controls25 (manuscript in preparation). Addition of CNM-Au8 to the media of human-induced pluripotent stem cell (iPSC)-derived motor neurones cocultured with iPSC-derived astrocytes from an ALS patient resulted in neuroprotection of the motor neurones from cytotoxic effects of the patient-derived astrocytes in a dose-dependent manner25 (manuscript in preparation). Taken together, these data support CNM-Au8 as a potential disease-modifying agent for the treatment of ALS.
Objectives
The primary objective is to assess the efficacy, safety, pharmacokinetics (PK) and pharmacodynamic (PD) effects of CNM-Au8 as a disease-modifying treatment for patients with ALS. Efficacy will be assessed as the change in motor neurone number as measured by neurophysiological electromyography assessments (eg, motor unit number index (MUNIX), motor unit size index (MUSIX), Split Hand Index (SI), Neurophysiology Index, MScanFit) and clinical endpoints. Exploratory clinical outcomes, while underpowered with this design, include change in ALSFRS-R score, the mean rate of change (ΔFS) of the ALSFRS-R from symptom onset, survival status, respiratory functions and composite disease progression.
Aim and study rationale
The aim of this trial is to assess the efficacy of CNM-Au8 in patients with ALS by utilising novel electrophysiologic markers of disease progression. ALS disease progression can be quantitatively assessed by MUNIX, which provides an index of the number of functional motor units, while potential reinnervation can be assessed by MUSIX.26 27 These measurements serve as neurophysiological markers of lower motor neurone loss in ALS.26 27 In a large multicentre clinical trial of subjects with ALS in the USA and Europe, MUNIX was demonstrated to be a reliable biomarker for clinical outcomes with an acceptable test–retest coefficient of variation, establishing MUNIX and MUSIX as potential paraclinical outcome measures for proof-of-concept clinical trials.26 27 Importantly, a longitudinal study of ALS patients demonstrated that MUNIX scores were more sensitive to decline over an 8-month period than ALSFRS-R scores, suggesting that MUNIX may be an earlier sensitive biomarker of motor neurone decline than the use of ALSFRS-R in clinical trials.28
This study uses electromyography to monitor motor neurone loss by MUNIX and to monitor reinnervation by MUSIX, respectively. MUNIX and MUSIX tests will be administered at Baseline and end-of-study (EOS, week 36). Clinical efficacy will be measured through change in the ALSFRS-R score, among other assessments. Results from this study will indicate whether CNM-Au8 is an effective disease-modifying treatment for patients with ALS, warranting further research in larger studies powered for clinical disease progression.
Methods and analysis
This is a multicentre randomised, double-blind, parallel group, placebo-controlled study comparing the efficacy, safety, PD and PK of CNM-Au8 in patients with ALS. Entry criteria require patients to be newly symptomatic within 24 months of screening and meet diagnostic criteria for ALS per the updated Awaji criteria.2 29
Test article and treatment administration
CNM-Au8 is an aqueous suspension of clean-surfaced, faceted nanocrystals consisting of gold (Au) nanoparticles produced under optimised growth conditions that lead to a preferred distribution of self-organised crystalline geometrical shapes (eg, hexagonal bi-pyramid, pentagonal bi-pyramid, tetrahedron, decahedron). Highly pure elemental Au nanocrystals are suspended in USP purified deionized water buffered with 6.5 mM sodium bicarbonate (NaHCO3), and nominally concentrated up to 0.5 mg/L (500 ppm). Patients will receive 30 mg of CNM-Au8, or placebo, once daily, first thing in the morning, by oral administration. CNM-Au8 or matching placebo will be administered in volumes of 60 mL from single-dose containers. All patients will receive their randomised oral treatment daily over 36 consecutive weeks during the double-blind treatment period.
Study design
This randomised, double-blind, placebo-controlled study is designed to demonstrate whether increasing central nervous system (CNS) bioenergetic cellular support in early symptomatic ALS patients with CNM-Au8 treatment can help preserve motor neurone survival and function. The primary endpoint is the mean change in the average difference between active treatment and placebo from baseline through week 36 for the MUNIX score, which quantitatively reflects the number of functional lower motor neurones in ALS.
In total, 42 sALS patients, meeting the inclusion and exclusion criteria (table 1), will be recruited from two multidisciplinary ALS expert clinics with significant clinical neurophysiology expertise:30 the Brain and Mind Centre, and Westmead Hospital in Sydney, Australia. Per protocol, all patients will be randomised (1:1) to receive CNM-Au8 (30 mg) or placebo daily over 36 consecutive weeks during the double-blind treatment period. Block randomisation will be completed by an independent statistician not available to the site investigators, study staff. Unblinded study personnel (eg, pharmacists) at each site will have access to the site-specific randomisation schedule, which are sequentially assigned. All site personnel delegated tasks related to study collection or assessment conduct are blinded to treatment allocation for the entirety of the trial. Participants may be unblinded by the study pharmacist in the case of a safety event after discussion with the medical monitor and the sponsor’s safety representative. Following the end of the double-blind treatment period, patients may be transitioned into an open-label extension study under a separate study protocol. Patients who elect to not transition into the open-label extension study will complete an EOS safety assessment 4 weeks following study drug discontinuation. The study scheme is shown in figure 1.
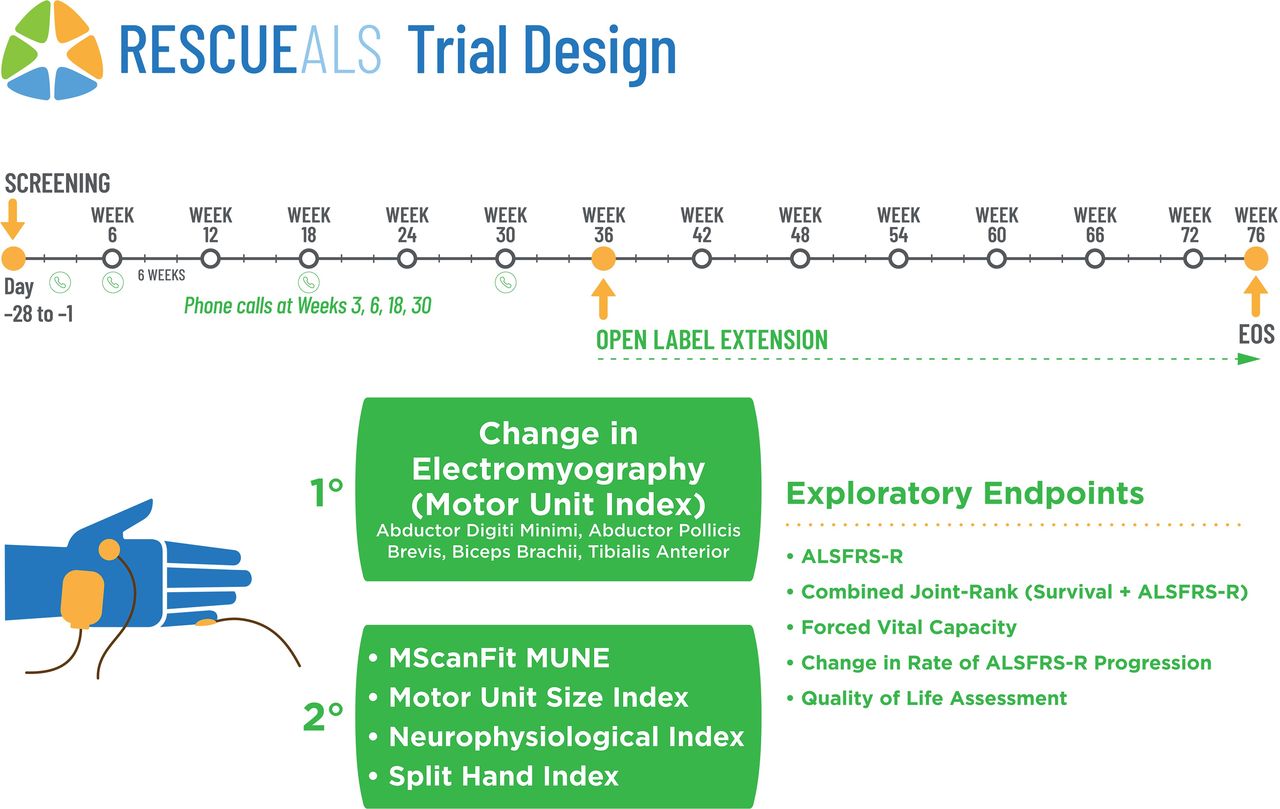
{kind=link}
Design of the study. Open circles on the timeline represent clinical visits or tele-visits as indicated. ALSFRS-R, ALS Functional Rating Score-Revised; MUNE, motor unit number estimation.
Inclusion and exclusion criteria
The complete estimated study duration will be approximately 70 weeks from the first patient-first visit to the last patient-last visit. The number of randomised patients is 42, with projected 36 evaluable, assuming an estimated 14% drop-out rate.
Patient and public involvement
To ensure that patient needs and experiences are taken into account, early in the design process, lead clinicians who specialise in ALS and who see ALS patients regularly, as well as a representative from the US-based non-profit, patient-focused ALS Association, were consulted. ALS patients were not directly involved in the design of this study.
Eligibility criteria
Study population will include patients with a probable laboratory-supported, clinically probable or definite ALS per the updated Awaji criteria as determined by a neurologist subspecialising in ALS.29 Table 1 defines the study inclusion and exclusion criteria.
The primary efficacy endpoint is the mean change in the difference between active treatment and placebo groups from baseline through week 36 in the averaged MUNIX scores. MUNIX studies will be carried out by trained neurophysiologists who have utilised and previously published on the technique, and will be performed in accordance with well-established methods, which includes having the same evaluator at each site assess the same study participants at each clinical visit.30 31 To independently ensure data quality, accuracy and interpretation of collected electromyography values, investigators at each site will evaluate the blinded data collected from the sister site prior to confirming the data for entry into the password-protected clinical trial database.
MUNIX will be performed on the following nerves assessed unilaterally on the least affected side: (i) ulnar nerve-abductor digiti minimi (ADM) muscle; (ii) median nerve-abductor pollicis brevis (APB) muscle; (iii) musculocutaneous nerve-biceps brachii (BB) muscle and (iv) common peroneal nerve-tibialis anterior (TA) muscle. The average change of the ADM, APB, BB and TA, the MUNIX(4) score, will be assessed every 12 weeks and analysed based on change from baseline to week 36.26 Changes in MUNIX and MUSIX values will be calculated as the percent change from baseline for each nerve indexed to 100%.
Secondary efficacy endpoints will involve additional neurophysiological assessments, including the following:
MScanFit motor unit number estimation of the APB will be undertaken by stimulating the median nerve at the wrist in accordance with established techniques.30–34
Neurophysiological index (NI) will be calculated form the ulnar nerve and recording from ADM muscle.35 The following formula will be used:
NI=CMAP (mV)* F-wave frequency (%)/Distal Motor Latency (ms), where CMAP is the compound muscle action potential
SI will be calculated using previously reported methodology.36 The SI will be calculated using the following formula:
SI= CMAP APB *CMAP FDI / CMAPADM
Exploratory efficacy endpoints will include the following:
The proportion of patients experiencing a >6-point decline in the ALSFRS-R.
The Combined Assessment of Function and Survival.
Overall survival.
The proportion of patients experiencing ALS clinical composite disease progression defined as the occurrence of death, tracheostomy, use of non-invasive ventilatory respiratory support, insertion of a percutaneous endoscopic gastrostomy tube or a 6-point drop in the ALSFRS-R score.
Changes in the rate of disease progression defined as the average change in the ΔFS score will be calculated as follows: Max ALSFRS-R minus current ALSFRS-R score/symptom duration in months.37
Changes in respiratory function as measured by forced vital capacity (FVC).
ALS-specific Quality of Life (ALSSQOL)-Short Form questionnaire.
Clinician’s global impression (CGI).
Patient’s global impression (PGI).
Differences in the proportion of patients utilising health economic outcome measures.
Time and event schedule
Study procedures for clinical visits will be performed as detailed in the time and event schedule (table 2). In addition, phone call tele-visits with each participant will occur at 6-week intervals to determine safety and collect concomitant medication information.
RESCUE-ALS clinical visit schedule
Screening visit (visit 1, up to 4 weeks prior to baseline visit)
Patients will be screened for the study after signing an approved informed consent form (ICF), which will be collected by the clinical site, and after ensuring inclusion/exclusion criteria are met. The ICF includes permission for collection and analyses of biological specimens (a copy of the ICF is provided as online supplemental material). Physical and neurological examinations will be performed, and vital signs, height, weight, body mass index (BMI) will be recorded. An electrocardiogram (ECG) will be performed. The visit will also involve an initial assessment of ALSFRS-R, FVC and the Columbia-Suicide Severity Rating Scale (C-SSRS). Prior/ongoing medications will be recorded. All patients will undergo laboratory evaluation of haematological and biochemical parameters; hepatitis B, C and HIV serology and urinalysis.
Supplemental material
Baseline visit (visit 0, week 0)
At the baseline visit, all patients will repeat the review of the inclusion/exclusion criteria. At the clinic, eligible patients will be randomised into active group (CNM-Au8) or control group (placebo) in a 1:1 ratio and the investigational product will be dispensed. This study visit includes baseline electromyography assessments (eg, MUNIX, MUSIX, CMAP and MScanFit) and FVC measurements. Scores on the following scales will be recorded: ALSFRS-R, PGI, CGI, ALSSQOL-SF, C-SSRS and a health utilisation form (for PEG, NIV, wheelchair use). Whole blood, plasma and urine will be collected for PD analyses. All patients will undergo physical and neurological examinations, as well as laboratory evaluation of haematological parameters and urinalysis. Vital signs, height, weight, BMI will be recorded. An ECG will be performed. Prior/ongoing medications will be recorded. In addition, women of childbearing potential will undergo a urine pregnancy test.
Treatment period (tele-visit 1) at week 3, and tele-visits weeks 6, 18 and 30
Phone call tele-visits with each participant occur at weeks 3, 6, 18 and 30 (not shown in table 2) to determine safety and collect concomitant medication information.
Treatment period (visits 2–4), weeks 12, 24 and 36
Patients will return to the clinic at weeks 12, 24 and 36 for electromyography and FVC measurements. Scores on the following scales will be recorded: ALSFRS-R, PGI, CGI, ALSSQOL-SF, C-SSRS and a health utilisation form (for PEG, NIV, wheelchair use). A Falls Questionnaire will be administered. Whole blood, plasma and urine will be collected for PD analyses, and whole blood will also be collected for PK analyses. All patients will undergo physical and neurological examinations, as well as laboratory evaluation of haematological parameters and urinalysis. Vital signs, height, weight and BMI will be recorded. An ECG will be performed. Prior/ongoing medications will be recorded. In addition, women of childbearing potential will undergo a urine pregnancy test.
End of study (visit 5) week 40
After completing week 36 procedures, all patients will return to the study site at week 40 for the EOS visit (4 weeks after the end of treatment visit). By the end of treatment, patients may elect to be transitioned into an open-label extension period in a separate study protocol. Patients who choose not to transition into the open-label extension study will complete an EOS safety assessment 4 weeks following study drug discontinuation. Patients who discontinue the study early will be asked to return to the study site for the early termination procedures 4 weeks after their last dose of active drug/placebo. At this visit, blood will be collected for laboratory evaluation of haematological parameters. Prior/ongoing medications will be recorded and a check for any adverse events will be performed. The C-SSRS will also be administered.
Safety monitoring
An independent Data Safety Monitoring Board (DSMB) will be responsible for monitoring the safety of patients in the study. The DMSB will hold regularly scheduled meetings on a quarterly basis. Additional ad hoc meetings may be held at the option of the DSMB, investigators or sponsor. The DSMB will review all of the available safety data throughout study and may make recommendations on the conduct of the study, including, if appropriate, study termination. The functioning of the DSMB are detailed in a DSMB Charter.
Safety will be assessed based on the frequency of treatment emergent adverse events (TEAEs), serious adverse events (SAEs), deaths, discontinuations due to adverse events, Falls Questionnaire scores and the C-SSRS scores. Changes from baseline in clinical laboratory results and vital signs will be summarised by treatment group and time point. TEAEs will be coded using the medical dictionary for regulatory activities and tabulated in incidence tables by system organ class and preferred term per treatment group. TEAEs will be further summarised by maximum severity and relationship to the investigational medicinal product. All safety summaries will be descriptive. All trial participants who suffer harm from trial participation will be compensated per the standard Medicines Australia clinical trial research agreement.
Withdrawal or premature discontinuation
A patient may withdraw from the study at any time for any reason without prejudice to their future medical care by the physician or the institution. The investigator or sponsor may withdraw the patient at any time (eg, in the interest of patient safety). Any patient removed from the study will complete an end of treatment visit and remain under medical supervision until study discharge is medically acceptable.
If the investigational product is prematurely discontinued, regardless of the reason, the final study evaluations will be performed as completely as possible. The information will be collected in the electronic case report form until the safety follow-up visit 28±5 days after the last intake of study drug. Adverse events that occur within 28±5 days after the last dose of study drug will be followed up until resolution, if possible.
Patients will be discontinued from the study for the following medical and/or administrative reasons: (i) patient request, or at the request of the patient’s legally acceptable representative; (ii) pregnancy, breast feeding or repeat non-compliance with the scheduled pregnancy testing; (iii) the investigator judges that continuation of the study would be harmful to the patient’s well-being; (iv) a TEAE or SAE that limits the patient’s ability to continue the study or (v) treatment interruption for 35 days or more.
Patients may be discontinued from the study for the following medical and/or administrative reasons: (i) at the specific request of the sponsor and in liaison with the investigator (eg, obvious non-compliance, safety concerns); (ii) if any exclusion criterion applies during the treatment period; (iii) substantial non-compliance with planned study procedures or (iv) use of illicit drugs or other substances that may, in the opinion of the investigator, have a reasonable chance of contributing to toxicity or otherwise confound the study results. Patients who withdraw or are withdrawn following administration of the investigational product will be replaced under this protocol at the discretion of the sponsor and investigator.
Pharmacodynamic (PD) endpoints
Blood, plasma and urine will be collected at baseline and at weeks 12, 24 and 36 and stored for subsequent PD analyses. PD analyses are designed to include blood metabolomic markers of cellular bioenergetics (eg, redox coenzymes: nicotinamide adenine dinucleotide, reduced and oxidized forms (NAD+, NADH), nicotinamide adenine dinucleotide phosphate, reduced and oxidized forms (NADP+, NADPH); energetic coenzymes: adenosine triphosphate (ATP), adenosine diphosphate (ADP), adenosine monophosphate (AMP) and antioxidants: glutathione disulfide (GSSG) and glutathione (GSH)) in addition to untargeted metabolomic pathway analyses. PD analyses also include changes in urinary neurotrophin receptor P75ECD levels and serum neurofilament light chain as markers of disease progression. PD analyses will be specified in a separate PD analysis plan to be completed prior to database lock.
Pharmacokinetic (PK) endpoints
Gold concentration data in whole blood will be summarised with descriptive statistics by treatment group at each time point of collection. Pharmacokinetic analysis will first involve whole blood sample collection before administration of CNM-Au8 at week 12 and at week 24. At week 36, whole blood samples will be collected for measurement of concentrations of Au before and at 1, 2, 4 and 6 hours, immediately after administration of the last dose of the investigational drug product at the site. Week 36 data will be used to estimate an apparent Cmax and Tmax and area under the curve over the 24 hours dosing interval. PK data will be used to construct a composite whole blood concentration-time profile for the Q12 week visits during the randomised treatment period.
Concomitant medications
Except for acetaminophen (paracetamol), ibuprofen, naproxen and second-generation antihistamines (including fexofenadine, loratadine and cetirizine); patients may not take any new prescription medications, over the counter (OTC) or dietary supplements from 14 days prior to baseline through the EOS follow-up unless used to manage a TEAE. Should a new concomitant medication or dietary supplement be necessary to manage a TEAE, the patient should report the occurrence of the adverse event and the medication(s) they are taking to treat the adverse event to his/her study physician as soon as possible. ALS disease-specific medications (eg, riluzole) are required per the inclusion criteria. The investigator will make every effort to contact the medical monitor or sponsor representative prior to administration of a new concomitant therapy prescription or OTC) after randomisation, unless the concomitant therapy is needed immediately for patient safety.
Data analysis and statistical methods
The primary endpoint, the mean change in the MUNIX(4) score value, will be analysed using mixed model for repeated measures with treatment (weeks 12, 24 and 36) as fixed effects and baseline ΔFS and ALSFRS-R scores, as well as site of disease onset (bulbar vs limb onset) as covariates. Estimates of least-square means, SEs and 95% CIs will be presented by treatment group. For the active treatment versus placebo comparisons, the least-square mean difference, the SE of the difference and the 95% CI of the difference will be presented. The trial evaluation populations will include the Intent to Treat Set, consisting of patients for whom a randomisation number has been assigned; the Safety Analysis Set, consisting of patients who were randomised and receive at least 1 dose of investigational product; the Partial Analysis Set, consisting of all patients in the Safety Set with at least one postbaseline electromyography measurement and the Per Protocol Set, consisting of all patients in the Safety Set who have completed 36 weeks of treatment with all primary and secondary endpoints evaluable.
Sample size calculations
Based on a longitudinal study of MUNIX score decline in ALS patients,16 a 38.4% reduction in the averaged MUNIX score at 36 weeks with a common SD of 19.8% was assumed for the study population. At a 1:1 (CNM-Au8 30 mg: placebo) allocation, it is estimated that 36 patients would be required to power the study with 80% power and 5% statistical significance rate, in order to detect a slowing of decline between active and placebo groups at 36 weeks. The study is thus powered to detect a 19.2% reduction in averaged MUNIX score at 36 weeks in the active group versus the placebo. Assuming a 12.5% estimated non-evaluable rate, it is planned to assign 42 patients (21 active:21 placebo) to randomised treatment, excluding replacement subjects as applicable.
Discussion
This phase 2 study is assessing the efficacy, safety and PK/PD effects of CNM-Au8 as a disease-modifying agent for the treatment of patients with ALS. CNM-Au8 offers an innovative approach for potentially treating neurodegenerative diseases, such as ALS, for which there are few effective treatments at present. As the first therapeutic nanocatalyst in development for neurodegenerative diseases, CNM-Au8 poses several advantages over small molecule and biologic agent approaches: it has a unique multimodal mechanism of action that can address age and disease-related bioenergetic failure, oxidative stress and proteostasis dysregulation. Because it is not metabolised or degraded, it is expected to maintain repeated catalytic activity until excreted. It is orally bioavailable and crosses the blood–brain barrier. A successful outcome from this study will establish the direct treatment of cellular bioenergetic failure as a therapeutic target for ALS and will support the utility of using electromyography endpoints as valid paraclinical biomarkers for ALS disease progression.
Ethics and dissemination
This study has been registered with ClinicalTrials.gov under the identifier NCT04098406 and has been approved by the Human Research Ethics Committee of the Western Sydney Local Health District with the ethics reference 2019/ETH12107, and is conducted with the principles of the Declaration of Helsinki, the guidelines of Good Clinical Practice and all other applicable regulations. The first study participant was enrolled on 16 January 2020. Protocol modifications, except those intended to reduce immediate risk to study patients, will be made by sponsor-initiated amendment. Institutional review board (IRB)/Human Research Ethics Committee (HREC) approval will be obtained before changes are implemented at the study sites.
Patients’ anonymity will be strictly maintained, and only coded patient data will be recorded on any form submitted to the sponsor and IRB/HREC. The site investigators will keep a patient log and individually identifiable protected health information (PHI) with the code key in a password-protected database.
Results of this study will be disseminated by presentations at scientific meetings and published in peer-reviewed scientific journal(s).
Ethics statements
Patient consent for publication
Acknowledgments
We thank, first and foremost, the study participants and their families for their support and willingness to engage in clinical research. We express our deep gratitude to the past and current members of our ALS Scientific Advisory Board: Drs Nicholas Maragakis (Johns Hopkins University School of Medicine), Jinsy Andrews (Columbia University), James Berry (Massachusetts General Hospital), Angela Genge (Montreal Neurological Institute and Hospital), Jonathan Glass (Emory University) and Piera Pasinelli (Thomas Jefferson University); as well as Dr Lucie Bruijn, former Chief Scientist and Vice President of Research of the ALS Association, for their advice and guidance in clinical trial design for ALS patients, their scientific feedback on our preclinical studies and their continued encouragement and support of our drug development efforts. We thank science writer W Todd Penberthy for assembling the study protocol into the initial draft of the manuscript and Mr Bjorn Pendleton of Pendleton Creative, LLC for creating and illustrating Figure 1.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @picturewing, @clenenano
Correction notice This article has been corrected since it first published. The provenance and peer review statement has been included.
Contributors The study was designed by collaboration among SV, MCK and MTH. MTH wrote the protocol with AR, RG, KSH, SV and MCK. PM and WH are the study site lead investigators. All authors contributed to the writing, editing and preparation of the final manuscript.
Funding Substantial study support was provided by a research grant from FightMND (https://fightmnd.org.au/), which is gratefully acknowledged. Additional support was also provided by Clene Nanomedicine. Support from the National Health and Medical Research Council of Australia (NHMRC) program grant (#1037746) and partnership project (#1153439) is also gratefully acknowledged. MK is supported by an NHMRC Practitioner Fellowship (#1156093). As the study sponsor, Clene Nanomedicine will have access to the final trial data set and will have authority over the following activities: study design; collection, management, analysis and interpretation of data; writing of the study report and the decision to submit the report for publication.
Competing interests AR, KSH, RG and MTH are full-time employees and hold stock or options in Clene Nanomedicine. SV and MK are directors of companies that hold equity in Clene Nanomedicine. SV, MK and their respective institutions receive research funding support for the RESCUE-ALS study from Clene Nanomedicine. MK is Editor-in-Chief of the Journal of Neurology, Neurosurgery and Psychiatry (BMJ Publishers, UK).
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.