Article Text

Abstract
Introduction Anaemia is a common complication of chronic kidney disease (CKD). Owing to the limitations of erythropoiesis-stimulating agents (ESAs), the current standard of care, there is a need to develop new therapies. Hypoxia-inducible factor prolyl-hydroxylase (HIF-PH) inhibitors might be a promising new treatment option. Molidustat is an oral HIF-PH inhibitor that stimulates the endogenous, predominantly renal, production of erythropoietin and was generally well tolerated in phase IIb clinical trials. Here, we report the design and rationale of two studies from the molidustat phase III programme: MolIdustat once dailY improves renal Anaemia By Inducing erythropoietin (MIYABI).
Methods and analysis MIYABI Non-Dialysis-Correction (ND-C) and MIYABI Non-Dialysis-Maintenance (ND-M) are randomised, open-label, parallel-group, multicentre studies that aim to demonstrate the efficacy of molidustat treatment compared with darbepoetin alfa in patients with anaemia and non-dialysis-dependent CKD. The secondary objectives are to assess the safety, pharmacokinetics and pharmacodynamics of molidustat treatment. MIYABI ND-C will recruit patients currently untreated with ESAs, whereas patients treated with an ESA will enter MIYABI ND-M. Each study will recruit 150 patients who will be randomised in a 1:1 ratio to receive either molidustat or darbepoetin alfa for 52 weeks, with efficacy evaluated during weeks 30–36. Study drug doses will be titrated regularly using an interactive voice/web response system with the aim of maintaining the patients’ haemoglobin (Hb) levels between ≥110 and <130 g/L. The primary objective will be achieved if, in molidustat-treated patients, the mean Hb level remains within the target range during the evaluation period, and if the change in the mean Hb level at evaluation time points from baseline is non-inferior to darbepoetin alfa.
Ethics and dissemination The protocols were approved by ethics committees at all participating sites. These studies will be conducted in accordance with the Declaration of Helsinki and the Good Clinical Practice guidelines. Results arising from these studies will be published in peer-reviewed journal(s).
Trial registration numbers NCT03350321; Pre-results, NCT03350347; Pre-results.
- renal anaemia
- chronic kidney disease
- molidustat
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
MolIdustat once dailY improves renal Anaemia By Inducing erythropoietin (MIYABI) Non-Dialysis-Correction (ND-C) and MIYABI Non-Dialysis-Maintenance (ND-M) are randomised controlled trials investigating the efficacy (30–36 weeks) and long-term safety (52 weeks) of molidustat for the treatment of renal anaemia, building on the results from earlier 16-week phase IIb studies.
In both studies, molidustat treatment will be compared with an erythropoiesis-stimulating agent (ESA) (darbepoetin alfa), the current standard of care for renal anaemia.
MIYABI ND-C and MIYABI ND-M will recruit patients who are currently untreated with ESAs and those who are treated with an ESA for at least 8 weeks before entering the study, respectively, allowing assessment of the efficacy of molidustat for both the correction and maintenance of haemoglobin levels and reflecting the patient populations observed in clinical practice.
The sample size of 150 patients per study is expected to be sufficient to address the efficacy objectives.
The open-label design is a potential limitation of both studies; however, they have been designed to minimise bias.
Introduction
Anaemia is a common complication of chronic kidney disease (CKD)1 that is associated with an increased risk of hospitalisation and mortality.2 3 The prevalence of anaemia associated with CKD (also known as renal anaemia) increases with increasing CKD stage, from 8.4% at stage 1 to 53.4% at stage 5 in the USA.1 In Japan, the reported prevalence of renal anaemia in patients with CKD stages 3–5 is 32.3%.4
Renal anaemia occurs predominantly due to erythropoietin (EPO) synthesis by the kidneys that is insufficient to maintain normal red blood cell levels.5 Treatment with an erythropoiesis-stimulating agent (ESA) is the standard of care for renal anaemia.6 ESAs effectively increase haemoglobin (Hb) levels, thereby reducing the necessity for blood transfusions and improving the health-related quality of life of patients7 8; however, their safety profile has some limitations. The use of high doses of ESAs, which are required to achieve and maintain adequate Hb levels in patients with CKD, has been associated with an increased risk of cardiovascular events and mortality.9–13 ESA therapy may also result in the development or worsening of hypertension14 and, in rare cases, cause antibody-mediated pure red cell aplasia (PRCA).15 In patients with cancer who experience anaemia, ESA use is associated with an increased thrombosis risk.16 Therefore, there is a need for new treatments for renal anaemia.
EPO production is regulated by oxygen-dependent hypoxia-inducible factors (HIFs).17 In normoxic conditions, HIF prolyl-hydroxylases (HIF-PHs) hydroxylate HIF-α subunits at two distinct proline residues, which results in their subsequent degradation by the proteasome. HIF-PHs are inactive in hypoxic conditions; therefore, HIFs are stabilised and induce the expression of several hypoxia-inducible genes, including EPO. 17 HIF-PH inhibitors, a new approach for the treatment of renal anaemia, stimulate production of endogenous EPO by the kidneys and liver.18 Several HIF-PH inhibitors are in clinical development and have been investigated in phase II trials in patients with non-dialysis-dependent CKD.19–22
In addition to elevating EPO levels, HIF-PH inhibitors may have other therapeutic effects due to regulation of expression of genes involved in iron metabolism. Patients with CKD commonly experience changes in iron metabolism resulting in functional iron deficiency, which contributes both to the development of renal anaemia23 24 and to hyporesponsiveness to ESA therapy.25–27 Consequently, patients with renal anaemia are often treated with intravenous or oral iron supplementation, as well as an ESA.6 Evidence suggests that HIF-PH inhibitors may increase the availability of iron for erythropoiesis, as demonstrated by reduced hepcidin levels in preclinical18 and clinical studies.19–21 28 However, there are potentially negative consequences arising from HIF-PH inhibition including pro-angiogenic effects, related to production of vascular endothelial growth factor (VEGF),17 and progression of CKD. Despite this, no safety signals have been reported from clinical trials of HIF-PH inhibitors and there have been no reported changes in VEGF levels.19–21
Molidustat, an oral HIF-PH inhibitor, mimics the physiological response to tissue hypoxia, thereby stimulating endogenous EPO production, predominantly by the kidneys.18 In preclinical studies, molidustat treatment resulted in dose-dependent production of EPO in healthy rats and cynomolgus monkeys.18 Molidustat prevented or reversed the decline of haematocrit in rodent models of renal anaemia and inflammation-induced anaemia, respectively, and almost normalised systolic blood pressure in a rodent model of CKD.18 Importantly, EPO levels were within the normal physiological range following molidustat treatment,18 whereas supraphysiological peak serum levels have been observed in patients receiving ESAs.29 Furthermore, owing to its mechanism of action, molidustat is unlikely to induce PRCA, which is caused by neutralising antibodies that recognise ESAs and endogenous EPO.30
The use of molidustat in patients with renal anaemia and non-dialysis-dependent CKD has previously been investigated in two phase IIb studies.22 In a double-blind, fixed-dose trial conducted in patients who had not previously been treated with an ESA, treatment with molidustat was associated with dose-dependent increases in Hb levels compared with placebo. In an open-label, active-comparator, dose titration trial, patients who received molidustat at starting doses of 25 or 50 mg once daily demonstrated similar Hb levels to those observed in patients who continued treatment with darbepoetin alfa. Molidustat was generally well tolerated over the 16-week treatment period in these trials.
The molidustat phase III programme entitled MolIdustat once dailY improves renal Anaemia By Inducing EPO (MIYABI) will consist of five studies, including three randomised controlled trials and two single-arm trials that will be conducted in Japan. In this article, we report the design and rationale of two of these studies, MIYABI Non-Dialysis-Correction (ND-C) and MIYABI Non-Dialysis-Maintenance (ND-M), which will investigate the efficacy (weeks 30–36) and long-term safety (up to 52 weeks) of molidustat for the treatment of renal anaemia.
Methods and planned analyses
Study designs, objectives and populations
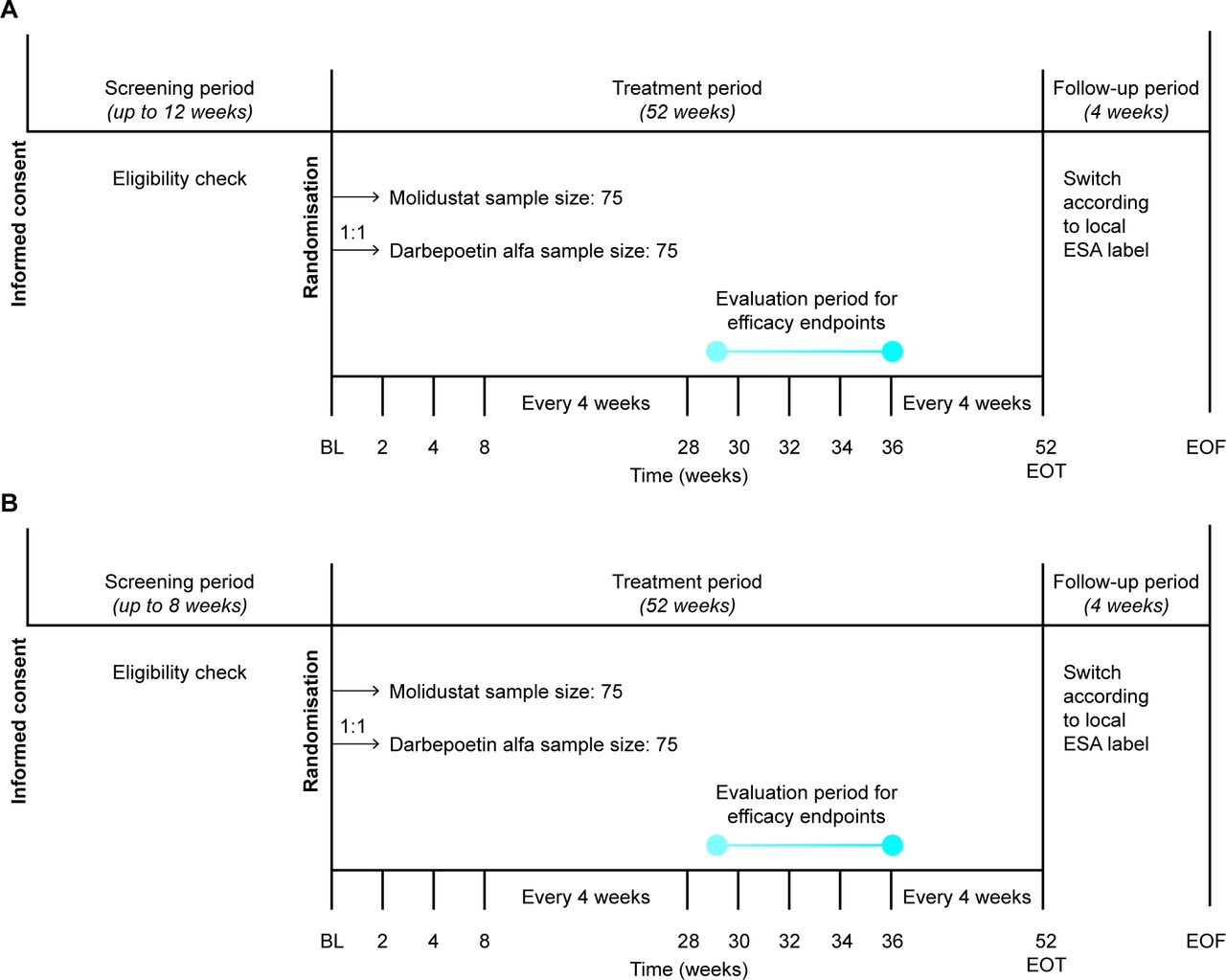
Both MIYABI ND-C and MIYABI ND-M are randomised, open-label, active-controlled, parallel-group, multicentre, phase III studies that will be conducted in Japan (figure 1). Both studies were initiated in December 2017 and have finished recruiting. MIYABI ND-C and MIYABI ND-M are expected to finish in October 2019 and November 2019, respectively. The primary objective of both studies is to demonstrate the efficacy of 30–36 weeks of molidustat treatment for renal anaemia in patients with non-dialysis-dependent CKD. The secondary objectives are to evaluate the safety and tolerability of 52 weeks of molidustat treatment and to evaluate its pharmacokinetic and pharmacodynamic parameters.
{kind=link}
Trial designs for (A) MolIdustat once dailY improves renal Anaemia By Inducing erythropoietin (MIYABI) Non-Dialysis-Correction and (B) MIYABI Non-Dialysis-Maintenance. Haemoglobin level and safety will be assessed at each study visit, conducted at the time points shown. BL, baseline; EOF, end of follow-up; EOT, end of treatment; ESA, erythropoiesis-stimulating agent.
In both studies, the study populations consist of adults aged 20 years or older with an estimated glomerular filtration rate (eGFR) of <60 mL/min/1.73 m2 (CKD stages 3–5) who are not undergoing dialysis and are not expected to start dialysis during the study period. Recorded Hb levels must be within prespecified levels during the respective screening periods to be eligible. Hb measurements must be taken at least 2 days apart, the difference between the two measurements must be <12 g/L and the last measurements must be taken within 14 days before randomisation. The main inclusion and exclusion criteria are shown in table 1, and the full list of exclusion criteria are detailed in online supplementary table 1.
Supplemental material
An overview of all inclusion criteria and key exclusion criteria
Eligibility for MIYABI ND-C was assessed during the screening period of up to 12 weeks. To be eligible, patients must be untreated with any ESAs in the 8 weeks before randomisation and have a mean of the last two Hb levels between ≥80 and <110 g/L. Eligibility for MIYABI ND-M was assessed during the screening period of up to 8 weeks. To be eligible, patients must be treated with the same ESA for at least 8 weeks before the screening period and have a mean of all Hb levels between ≥100 and <130 g/L.
In each study, approximately 150 eligible patients will be randomised in a 1:1 ratio to receive either molidustat or darbepoetin alfa over a 52-week treatment period. Allocation to treatment arms will be achieved using an interactive voice/web response system (IxRS) at the baseline visit. In MIYABI ND-C, randomisation will be stratified by medical history of thromboembolic events (yes or no for myocardial infarction, pulmonary thromboembolism, stroke (excluding haemorrhagic stroke) or acute limb ischaemia) and by mean Hb level at screening (<90 or ≥90 g/L). In MIYABI ND-M, randomisation will be stratified by medical history of thromboembolic events and by previous ESA dose group (low or high).
The safety of each study will be overseen by a data monitoring committee consisting of independent clinical experts and an independent biostatistician, supported by an independent statistical analysis centre, whose main role will be to recommend a change, interruption or termination of the study if a safety signal is detected.
Treatments
In MIYABI ND-C, molidustat will be administered orally at a starting dose of 25 mg once daily and darbepoetin alfa will be administered by subcutaneous injection at a starting dose of 30 µg every 2 weeks. A dose adaptation visit will occur after the first 4 weeks of treatment to avoid excessive elevation of Hb level after the initiation of treatment (online supplementary table 2).
In MIYABI ND-M, molidustat will be administered orally at a starting dose of 25 or 50 mg once daily, depending on the previous ESA dose received (online supplementary table 3). The starting dose of darbepoetin alfa in MIYABI ND-M will be determined based on the previous dose of ESA received.
In both studies, doses of molidustat or darbepoetin alfa will subsequently be titrated in a stepwise manner at scheduled regular dose control visits using IxRS, with the aim of correcting and maintaining (MIYABI ND-C) or maintaining (MIYABI ND-M) the Hb levels of patients within the desired range (≥110 to <130 g/L; online supplementary table 4). Planned doses for the titration of molidustat are 5, 12.5, 25, 50, 75, 100, 150 and 200 mg once daily. Planned doses for the titration of darbepoetin alfa are 15, 30, 60, 90, 120 and 180 µg every 2–4 weeks.
In MIYABI ND-C, molidustat dose control visits will be scheduled for every 4 weeks, starting after 8 weeks of treatment, whereas darbepoetin alfa dose control visits will initially occur every 2 weeks. In MIYABI ND-M, molidustat dose control visits will be scheduled for every 4 weeks, starting after 4 weeks of treatment. Darbepoetin alfa dose control visits will occur every 2–4 weeks, according to previous ESA dose and dose interval. In both studies, after 8 weeks of treatment, if a patient is receiving a darbepoetin alfa dose once every 4 weeks then dose control visits will also occur every 4 weeks.
In both studies, in cases of excessive elevation of a patient’s Hb level (rate of Hb rise >10 g/L per 2 weeks or >20 g/L per 4 weeks) during the treatment period, investigators may decrease the dose of molidustat or darbepoetin alfa at any time. If the administered dose of molidustat or darbepoetin alfa is the minimum dose step, the dose may be suspended.
Iron, vitamin B12 and folate supplementation is permitted during each study if required and will be administered according to Japanese guideline recommendations.31 Iron supplementation will be orally administered to reach a target serum ferritin level of at least 100 ng/mL or transferrin saturation of at least 20%.
For both studies, further ESA treatment may be initiated at the discretion of the investigator after the final administration of the study drug. Details of the ESA treatment regimen will be recorded if treatment is initiated in the 4-week follow-up period.
Variables
Variables will be collected from the screening period to 4 weeks after the 52-week treatment period. In both studies, the primary efficacy variables are the mean Hb level during the evaluation period (weeks 30–36) and its change from baseline. Safety variables will include adverse events (AEs), vital signs and laboratory and clinical parameters, including observations from ECG and ophthalmological examinations.
Exploratory variables will include measures of renal function, iron metabolism and health-related quality of life assessments. Full descriptions of secondary efficacy variables, safety variables and exploratory variables are provided in box 1.
Study variables
Secondary efficacy variables:
Responder rate, defined as the proportion of responders among the patients in the analysis set.*
Proportion of patients who meet each component of the response.*
Hb level and change from baseline (measurement at each visit and mean during the evaluation period).
Proportion of patients whose mean Hb level is in, above or below the target range during the evaluation period.†
Proportion of patients whose Hb level is in, above or below the target range at each visit.
Proportion of patients whose maximum rise in Hb level between each consecutive visit is above 5 g/L/week.‡
MIYABI ND-C-specific secondary efficacy variables:
Rate of rise in Hb level (g/L/week) at the first-dose change up to week 8.§
Rate of rise in Hb level (g/L/week) at the first-dose change up to week 4.
Cumulative proportion of patients who achieve the lower limit of the target Hb range at least once at each visit.
Safety variables:
Adjudicated adverse events.¶
Adverse events including serious adverse events.
Change in vital signs (pulse rate and blood pressure).
12-Lead ECG parameters.
Observations of ophthalmological examination (fundus and anterior ocular segment examination and intraocular pressure measurement).
Laboratory examinations (including haematology, coagulation, clinical chemistry, electrolyte, HbA1c, PTH and TSH).
Exploratory variables:
Conversion to end-stage renal disease (eGFR <15 mL/min/1.73 m2)
Parameters of renal function: cystatin C, serum creatinine, eGFR and urine albumin-to-creatinine ratio.
Parameters of iron metabolism.
EQ-5D-5L.
TSQM.
Vascular endothelial growth factor.
*Response is defined by the following three components: (i) mean of Hb levels during the evaluation period is within the target range; (ii) ≥50% of the Hb levels during the evaluation period are within the target range; (iii) did not receive rescue treatment up to the end of the evaluation period. A responder is an individual who meets all three components of the response.
†The patient must have at least two of the four Hb levels during the evaluation period.
‡Defined as change in Hb level/duration between two visits (weeks).
§Rate of rise in Hb level is derived as change in Hb level from baseline to the first-dose change of the study drug (including the change in interval of administration of darbepoetin alfa) up to week 8 divided by duration of the starting dose (in weeks). If no dose change is performed up to week 8, then an Hb level at week 8 and the date of week 8 visit will be used for the calculation of change in Hb level and duration. If no valid Hb level (collected at scheduled visit and assessed by the central laboratory) is available at the visit of the first-dose change, the following postbaseline Hb level will be used for imputation in the hierarchical order until one valid determination is available: (1) local Hb level at the visit (ie, visit of the first-dose change or week 8), (2) the latest central Hb level before the visit, (3) the latest local Hb level before the visit. For patients with no available Hb level, Hb level of 80 g/L will be used for the imputation as ‘worst-case’ approach given the possible worst value.
¶Adjudicated adverse events include death, myocardial infarction, unstable angina pectoris, stroke or transient ischaemic attack, pulmonary thromboembolism or acute limb ischaemia.
eGRF, estimated glomerular filtration rate; EQ-5D-5L, EuroQol 5-dimension 5-level questionnaire; Hb, haemoglobin; HbA1c, glycated haemoglobin; MIYABI, MolIdustat once dailY improves renal Anaemia By Inducing erythropoietin; ND-C, non-dialysis-correction; PTH, parathyroid hormone; TSH, thyroid-stimulating hormone; TSQM, treatment satisfaction questionnaire for medication.
In both studies, to investigate systematic exposure to molidustat, as well as the relationship between molidustat exposure and response, sparse sampling from all patients will be conducted for pharmacokinetic and pharmacodynamic parameters.
Quality assurance and data management
For both studies, audits may be conducted by a member of the sponsor’s quality assurance unit to assess the performance of the study at any of the study sites. In addition, sites may be inspected by regulatory health authority representatives, independent ethics committees and institutional review boards.
For both studies, data will be recorded by investigational site personnel onto the validated and password-protected electronic data capture system Rave (Medidata Solutions). All records identifying the patient will be kept confidential and will not be made available either to the public or the sponsor. All personal information made available for inspection will be handled in strictest confidence and in accordance with local data protection laws. Data will be pseudonymised for analysis. The sponsor will have access to the full trial dataset.
The sponsor maintains clinical trial insurance coverage for each of the studies to provide compensation in the unlikely event that a patient is harmed from participation in any of these clinical trials.
Statistical analysis
Data will be analysed for each study, and data from both studies will also be combined in an integrated analysis. All variables will be analysed descriptively with appropriate statistical methods including categorical variables by frequency tables and continuous variables by summary statistics.
For both studies, the primary analysis set for efficacy will be the full analysis set (FAS), which will include all randomised patients who have at least one baseline Hb level (ie, at least one Hb level measured before the first dose of the study drug) and will be analysed as randomised. Additionally, the per-protocol set will include all patients from the FAS who have at least two Hb levels measured in the evaluation period (weeks 30–36); this set will be used for sensitivity analyses in efficacy and analysed as treated.
For both studies, the primary objective will be achieved if two conditions are met. The first condition is that the mean Hb level during the evaluation period (weeks 30–36) in the molidustat treatment arm is within the target range (≥110 to <130 g/L). The mean Hb level during the evaluation period in the molidustat treatment arm will be calculated using the mean Hb levels of each patient. If the lower limit of the two-sided 95% CI of the mean of the molidustat group is greater than or equal to the lower limit of target Hb level (ie, ≥110 g/L) and if the upper limit of the two-sided 95% CI is less than the upper limit of target Hb level (ie, <130 g/L), it will be established that the mean Hb level of the molidustat group is within the target range. The two-sided 95% CI will be estimated using one sample t-statistics. The second condition is that molidustat is non-inferior to darbepoetin alfa. This will be established if the lower limit of the two-sided 95% CI for the difference in the change in mean Hb level during the evaluation period from baseline (molidustat minus darbepoetin alfa) is above –10 g/L with a non-inferiority margin of 10 g/L. This margin was chosen in line with guidelines that specify that a variation of approximately 10 g/L is considered acceptable in clinical practice.31 The difference in change between the treatment groups and its two-sided 95% CI will be estimated using an analysis of covariance model, including treatment group (and previous ESA dose group in MIYABI ND-M; low/high) and previous thromboembolic events (yes/no) as fixed effects and baseline Hb level as a covariate.
Several descriptive and exploratory subgroup analyses are planned for both studies, including age, sex, baseline weight, prior thromboembolic event, baseline eGFR, main cause of CKD and duration of CKD. Baseline Hb level will also be included as a subgroup in MIYABI ND-C and previous ESA dose group will be included in MIYABI ND-M.
All patients who receive at least one dose of the study drug will be included in the safety analysis and will be analysed as treated. The incidence of treatment-emergent AEs will be tabulated using medical dictionary for regulatory activities terms and additional tables produced for serious and/or drug-related treatment-emergent AEs. Adjudicated AEs will also be summarised descriptively.
Determination of sample size
The sample size of 75 patients per treatment arm in each study was chosen to obtain sufficient data for assessment of the long-term safety of molidustat (at least 100 patients treated with molidustat for 1 year), when data from MIYABI ND-C and MIYABI ND-M are combined, assuming that 25% of patients will prematurely stop the study medication.
For the primary efficacy analysis, a sample size of 75 patients per treatment arm has >98% power to establish that the mean Hb level during the evaluation period is within the target range, assuming an SD of 10–13 g/L based on previous phase IIb studies. This sample size is also sufficient to reject the null hypothesis with 90% power that molidustat is inferior to darbepoetin alfa, with a non-inferiority margin of 10 g/L at a one-sided 2.5% significance level, assuming the expected difference between molidustat and darbepoetin alfa to be 0 g/L and a common SD of 10–13 g/L.
Patient and public involvement
Patients are not involved in the design and conduct of the studies.
Discussion
Previous preclinical studies and clinical trials have shown promising results for the efficacy and safety of molidustat for the treatment of renal anaemia.18 22 The MIYABI ND-C and MIYABI ND-M phase III studies are expected to build on previous phase IIb studies and demonstrate the efficacy and safety of molidustat compared with darbepoetin alfa for the correction and maintenance of Hb levels in patients with non-dialysis-dependent CKD. The studies are randomised, open-label, active-controlled, parallel-group, multicentre trials encompassing approximately 300 patients (150 patients per study). This sample size is expected to be sufficient to determine whether the mean Hb level during the evaluation period (weeks 30–36) in the molidustat treatment arm is within the target range, to demonstrate the non-inferiority of molidustat to darbepoetin alfa using a non-inferiority margin of 10 g/L and to assess the long-term safety of molidustat treatment. However, it should be noted that this sample size is too small to adequately assess safety with regard to the risk of cardiovascular events. Previous phase IIb studies investigated up to 16 weeks of molidustat treatment,22 whereas the total treatment period in these phase III studies is 52 weeks, with efficacy evaluated after 30–36 weeks of treatment. Furthermore, MIYABI ND-C will be the first clinical trial to compare molidustat treatment with an active comparator for the correction of Hb levels in patients with non-dialysis-dependent CKD, having previously been tested against placebo in a phase IIb study.
Several exploratory variables will also be investigated during the trials in order to assess other potential effects of HIF-PH inhibition. Measures of iron metabolism are expected to provide further evidence regarding the availability of iron for erythropoiesis following molidustat treatment.18 28 Assessment of VEGF levels and ophthalmological examination will be conducted to assess the theoretical risk of VEGF-mediated diabetic retinopathy. Furthermore, measures of renal function including cystatin C, serum creatinine, eGFR and urine albumin-to-creatinine ratio will be monitored throughout the study to determine whether molidustat treatment influences the progression of CKD and conversion to end-stage renal disease.
Both studies will be conducted using an open-label design; however, owing to the recognised limitations, they have been designed to minimise the potential introduction of any bias and to ensure the results are robust. First, objective Hb levels have been specified as an efficacy endpoint. Second, in any treatment group, the next dose will be determined by IxRS in accordance with the prespecified dosing protocol, rather than by individual investigators. Third, sufficient supplementation of vitamin B12, folic acid and serum iron will be confirmed before the first study treatment to reduce the impact of supplementation after study treatment. Finally, health-related quality of life questionnaires will be administered before a patient is told their current Hb level to minimise any influence on outcomes.
MIYABI ND-C and MIYABI ND-M are part of a larger molidustat phase III clinical development programme conducted in Japan that also includes a randomised, active-controlled, double-blinded trial (MIYABI HD-M) and two single-arm studies (MIYABI HD-C, MIYABI PD) that will investigate the efficacy and safety of molidustat in patients with CKD on haemodialysis and peritoneal dialysis. The design and rationale of these three studies are presented in a companion article.32 In total, the five phase III studies will include approximately 600 participants and will reflect a broad clinical spectrum of patients with anaemia and CKD.
Ethics and dissemination
The studies are being conducted in accordance with the Declaration of Helsinki and the Good Clinical Practice guidelines.
Informed consent was obtained from patients by the site investigator or a designated person before entering the studies and may be withdrawn at any time. Proposed protocol amendments must be agreed by the sponsor and investigators and approved by independent ethics committees and institutional review boards. Protocol amendments must be signed by the principal investigator and have received all external approvals before coming into effect at the respective centre. If there is a change in the protocol that necessitates a change in consent, the investigator will inform patients of the changes in a timely manner and ask each patient to reconfirm their participation in the study by signing a revised consent form.
Both MIYABI ND-C (NCT03350321) and MIYABI ND-M (NCT03350347) have been registered on ClinicalTrials.gov. Results arising from these studies will be published in peer-reviewed journal(s), but there are no plans to publicly release the full protocol, participant-level dataset or statistical code from either of the studies.
Acknowledgments
Medical writing support was provided by Cerys Evans, PhD, of Oxford PharmaGenesis, Oxford, UK. Ken Miyazaki, Eriko Ogura and Hitomi Mizutani of Bayer Yakuhin Ltd reviewed the manuscript for statistical and/or scientific accuracy.
References
Footnotes
Contributors HY, TA and TY contributed to the design of these studies and the development of the original study protocols. MT contributed to drafting and revising the manuscript. HY, TA, MT and KI contributed to the critical revision of the article for intellectual content. YM contributed to development of the statistical analysis plan and assisted in the preparation of the manuscript. All authors approved the final version of the manuscript and agreed to be accountable for all aspects of the work, ensuring that questions related to the accuracy or integrity of any part of the work are appropriately resolved.
Funding These studies are funded by Bayer Yakuhin Ltd. The trials were designed and are being conducted by employees of Bayer Yakuhin Ltd, in consultation with healthcare professionals including TA and HY. Bayer Yakuhin Ltd is responsible for the design of these studies and the analysis and interpretation of data collected by investigators. Bayer Yakuhin Ltd and participating contract research organisations are responsible for management of data and writing the report. Bayer Yakuhin Ltd will make the final decision regarding submission of a manuscript for publication.
Competing interests MT, YM, KI and TY are employees of Bayer Yakuhin Ltd. TA received consulting and lecture fees from Bayer Yakuhin Ltd during the conduct of the study. TA has also received consulting, lecture or manuscript fees outside the submitted work from Astellas, GlaxoSmithKline, JT Pharmaceuticals, Kissei Pharmaceutical Co. Ltd, Kyowa Hakko Kirin, Nipro Corporation, Fuso Pharmaceutical Industries Ltd, Ono Pharmaceutical Co. Ltd, Chugai Pharmaceutical Co. Ltd and Torii Pharmaceutical Co. Ltd. HY received consulting fees from Bayer Yakuhin Ltd during the conduct of the study.
Ethics approval Documented approval from appropriate independent ethics committees and institutional review boards has been obtained. The MIYABI ND-C study has been approved by the institutional review board of Kyushu University Hospital (application number: 20171211), Nihon University Hospital (application number: 20171130) and a further 60 sites. The MIYABI ND-M study has been approved by the institutional review board of Kyushu University Hospital (application number: 20171211), Nihon University Hospital (application number: 20171130) and 59 additional sites.
Provenance and peer review Not commissioned; externally peer reviewed.
Patient consent for publication Not required.