Article Text

Abstract
Objectives To define a series of clinical trial transparency measures and apply them to large pharmaceutical and biotechnology companies and their 2014 FDA-approved drugs.
Design Cross-sectional descriptive analysis of all clinical trials supporting 2014 Food and Drugs Administration (FDA)-approved new drug applications (NDAs) for novel drugs sponsored by large companies.
Data sources Data from over 45 sources, including Drugs@FDA.gov, ClinicalTrials.gov, corporate and international registries; PubMed, Google Scholar, EMBASE, corporate press releases, Securities and Exchange Commission (SEC) filings and personal communications with drug manufacturers.
Outcome measures Trial registration, results reporting, clinical study report (CSR) synopsis sharing, biomedical journal publication, and FDA Amendments Acts (FDAAA) compliance, analysed on the drug level.
Results The FDA approved 19 novel new drugs, sponsored by 11 large companies, involving 553 trials, in 2014. We analysed 505 relevant trials. Per drug, a median of 100% (IQR 86%–100%) of trials in patients were registered, 71% (IQR 57%–100%) reported results or shared a CSR synopsis, 80% (70%–100%) were published and 96% (80%–100%) were publicly available in some form by 13 months after FDA approval. Disclosure rates were lower at FDA approval (65%) and improved significantly by 6 months post FDA approval. Per drug, a median of 100% (IQR 75%–100%) of FDAAA-applicable trials were compliant. Half of reviewed drugs had publicly disclosed results for all trials in patients in our sample. One trial was uniquely registered in a corporate registry, and not ClinicalTrials.gov; 0 trials were uniquely registered in international registries.
Conclusions Among large pharmaceutical companies and new drugs, clinical trial transparency is high based on several standards, although opportunities for improvement remain. Transparency is markedly higher for trials in patients than among all trials supporting drug approval, including trials in healthy volunteers. Ongoing efforts to publicly track companies’ transparency records and recognise exemplary companies may encourage further progress.
- clinical pharmacology
- health Policy
- law (see medical law)
- medical ethics
- public health
- bioethics
- pharma ethics
- clinical trial transparency
- good pharma scorecard
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
- clinical pharmacology
- health Policy
- law (see medical law)
- medical ethics
- public health
- bioethics
- pharma ethics
- clinical trial transparency
- good pharma scorecard
Strengths and limitations of this study
This analysis uniquely evaluates new drugs and large biotechnology and pharmaceutical companies on several different clinical trial transparency standards on the ethics and legal levels—not merely the usual crude measure of whether companies have reported results for the trials they registered on ClinicalTrials.gov. This analysis is limited to drugs approved by the Food and Drugs Administration (FDA) in 2014 for which the new drug application (NDA) holder was a large company. Subsequent and previous publications evaluate more drugs and trial sponsors.
Our review is global; we searched over 39 public trial registries, including international, corporate and patient registries, and we provide an assessment on the completeness of ClinicalTrials.gov and the value of allocating resources to link existing trial databases. A further innovation is rigorously validating study findings and data with NDA holders (pharmaceutical companies).
We created a company ranking to accompany the drug rankings on clinical trial transparency performance. We also analysed three different samples of trials: trials conducted in patients, all trials (including those involving healthy volunteers), and Food and Drug Administration Amendments Act (FDAAA) applicable trials.
This study captures static snapshots of clinical trial transparency for new drugs approved by the FDA in 2014, at the time of FDA approval, 3 months post FDA approval, 6 months post FDA approval and 13 months post FDA approval. Subsequent trial disclosures are not captured.
We do not count abstracts submitted to scientific conferences as ‘publications’ and we included only those trials related to the indication(s) for which the new drug was initially approved, in our legal compliance assessments.
Introduction
Despite its importance in supporting evidence-based patient care, prescription guideline development, formulary decisions, public trust in research and healthcare innovation, transparency around clinical trial results does not always occur.1–7 Despite the promulgation of multiple legal and guidance standards in the 20 years since Congress enacted the first law requiring registration of certain trials,8 transparency practices remain highly variable across research sponsors like universities and drug companies.4 7 9 10 Moreover, studies that measure the transparency of trials, drugs, drug manufactures and research sponsors often use markedly different transparency measures and standards, yielding different findings and progress reports. A research sponsor, trial or drug may look transparent in one study and opaque in another.
Efforts to establish clinical trial transparency standards and improve practices span decades. In 1997, the USA adopted the Food and Drug Administration Modernization Act8 requiring the registration of drug trials for serious or life-threatening conditions. In 2007, the Food and Drug Administration Amendments Act (FDAAA) expanded disclosure requirements to include trials for all types of health conditions and required that select trial results be publicly reported for FDA-regulated products.11 In 2008, the World Medical Association identified trial registration and reporting as an ethical obligation, for all trials, in the Declaration of Helsinki.12 A bevy of other leading scientific and development organisations have also endeavoured to improve research transparency, including the Gates Foundation, WHO and Wellcome Trust.13–20 Most recently, the US Department of Health and Human Services and National Institutes of Health (NIH) released policies and rules to further expand and clarify the types of trials for which reporting is required.21–23 The new NIH policy requires, for the first time, results reporting for all NIH-sponsored trials, including phase I trials conducted in healthy volunteers.24
These efforts have helped foster a culture of transparency in research, but they have also introduced ambiguous, and at times conflicting, standards. To help harmonise standards going forward, assess the current state of transparency, as well as to identify and reform areas where further improvement is needed, we defined a series of concrete transparency measures and applied them to drugs approved by the FDA in 2014 and their large company sponsors.
Our transparency standards include both measures of compliance with US legal requirements—that is, whether companies report what FDAAA requires them to—and two expanded standards applying to broader ranges of trials, one of which includes phase I trials in healthy volunteers, and the other that only looks at trials in patients. This paper is a continuation of an initiative, called The Good Pharma Scorecard, that began with benchmarking the transparency of drugs approved by the FDA in 2012, sponsored by large drug companies, on a significantly narrower set of measures.25–27
This analysis is innovative by uniquely evaluating new drugs and their sponsors on several different standards and measures—not merely the usual crude measure of whether companies have reported results for the trials they registered on ClinicalTrials.gov. Our review is global; we searched 39 public trial registries, including international, corporate and patient registries, and provide an assessment on the completeness of ClinicalTrials.gov. A further innovation is rigorously validating all study measures and data with NDA holders (pharmaceutical companies). Lastly, we added a company ranking to accompany the drug rankings.
Methods
Data
We used data from multiple (over 44) sources (details in the online supplementary appendix 1), including Drugs@FDA.gov, a publicly accessible database containing records of FDA regulatory decisions; 39 trial registries including ClinicalTrials.gov, the Aggregate Analysis of ClinicalTrials.gov database (AACT), individual corporate registries, the WHO’s International Clinical Trials Registry Platform, which aggregates 16 country registries, and the Clinical Study Data Request Repository; journals indexed in PubMed, Google Scholar and EMBASE; and corporate press releases. Additional information was obtained through personal communications with drug manufacturers. All databases were accessed several times in January–August 2016.
Supplementary file 1
Sampled drugs and companies
We examined clinical trials relating to new molecular entities (NMEs), and new combination drugs containing at least one NME component, that were approved by the FDA in 2014.28 We confined our analysis to new drug applications (NDA) that were sponsored by the 20 largest biotechnology and pharmaceutical companies, or their subsidiaries, as measured by 2014 market capitalisation ranking.29 We began with large companies to highlight practices among those with the most resources available to deploy toward satisfying transparency standards that are more comprehensive than those currently imposed by law. Subsidiaries and parent companies were identified by searching corporate websites, press releases and SEC filings and via communications with companies during the data validation process. Future versions of our scorecard will expand the analysis to include all trial sponsors, including small-sized and medium-sized companies and public sponsors.
Sampled clinical trials
A list of every clinical trial included in the NDA was created by reviewing the 2014 FDA approval packages for each of the 19 drugs. Basic characteristics of each trial were extracted (details in the online supplementary appendix 2). We excluded trials terminated without enrolment, expanded access trials, observational studies and trials that were ongoing or not at least 1 year past their primary completion date (PCD) by our study cut-off date of 1 February 2016. PCD was defined as on ClinicalTrials.gov. Observational studies (which constituted 5 of the 553 studies we reviewed) were generally excluded because they were ongoing at the time of our study cut-off date. Additionally, they are not covered under FDAAA legal requirements to report trial results.
From this ‘all-trials’ sample, we then selected two subsamples. The ‘trials-in-patients’ subsample was confined to trials conducted in patients (as opposed to healthy volunteers). The ‘FDAAA trials’ subsample was limited to trials subject to mandatory registration and results reporting under FDAAA. Because our analyses examine both legal compliance with FDAAA and satisfaction of a more aspirational standard, different samples of trials were required.
The legal requirements under FDAAA for reporting trial results apply only to ‘controlled clinical investigations(s), other than a phase I clinical investigation’30 of a drug that is the subject of an approved NDA or for which an NDA would be required in order for the drug to be legally marketed in the USA. The requirements apply only if the trial began after 27 September 2007 or was ongoing as of 26 December 2007. Finally, the trial must meet one of the following conditions: (1) at least one US site, (2) conducted under an FDA investigational new drug application, or (3) involve a drug, biologic or device manufactured in USA or its territories, and exported for research.11 30
Data collection methods
Search terms to match trials in the registries included the trial’s organisational identification number, product name, chemical name(s), number of participants and other characteristics captured from the FDA approval packages. We abstracted all available characteristics on each trial from the registry (details in the online supplementary appendix 2). We matched trials to journal articles using a minimum of three trial characteristics, and searched the registries for links to publications.
For each trial, data were retrieved by at least two research assistants who received training and worked independently (details in the online supplementary appendix 3). Discrepancies between coders’ findings were resolved by consensus. Final datasets and findings were sent to drug companies for validation (details in the online supplementary appendix 4). Feedback from companies was generally incorporated into findings if it could be validated through our public sources. For example, companies in some cases provided a web link to a publication missed in our matching process.
The study did not undergo Institutional Review Board (IRB) review because it was not human subjects research.
Outcome measures
Our first outcome measure examined whether the trials in our samples were registered in any public registry, including corporate and international registries. Second, we determined whether either trial results or a clinical study report (CSR) synopsis were provided in the registry. Third, we determined whether each trial was published in a journal indexed by PubMed, Google Scholar or EMBASE. Fourth, we deemed each trial ‘publicly available’ if it had results reported in a registry, a CSR synopsis provided in a registry or results published in a journal.
Results were considered available if received by a registry or published by 1 February 2016. This date was chosen to provide a generous period of time for reporting results: at least 1 year after FDA approval of the drug plus a 1-month grace period.
We also measured the availability of trial results at the time each drug was approved by the FDA, 3 months after approval and 6 months after approval to track reporting timelines. Additionally, we tracked where trials were registered, reported or had shared CSRs to get a sense of the overall use of corporate, national and international registries and the need to invest in linking multiple databases together.
We applied these measures to two different samples of trials (table 1): (1) all trials, including trials enrolling healthy volunteers and patients, and (2) only trials enrolling patients. The all-trials analysis evaluates companies against the World Medical Association12 recommendation that all trials be registered and have results disclosed to honour ethical obligations to research subjects, both healthy volunteers and patient participants, as described in the Declaration of Helsinki and Belmont Report. The Belmont Report defines research as, ‘an activity designed… to develop or contribute to generalizable knowledge’, which generally requires the dissemination of research results. In contrast, companies generally only commit to disclosing results for trials enrolling patients, not healthy volunteers, as expressed in their trade association codes of conduct. Therefore, we also applied our transparency standards to those trials that enrolled patients in the intent-to-treat population. That analysis excluded patients with renal or hepatic impairment (who did not have the condition being studied) and trials in healthy volunteers.
Transparency measures and analysed clinical trial samples
We also assessed the extent to which trials subject to FDAAA met that statute’s transparency requirements. Setting cut-off points for this analysis was complex because there is a disagreement among companies about what the statute requires. There is broad agreement that FDAAA requires trial registration within 21 days after enrolling the first participant (we gave sponsors a 7-day grace period to account for delayed postings, weekends, holidays and time zones). However, two views exist about when results must be reported, both of which are plausible. One interpretation, which we call the ‘trial completion date’ view, is that results generally must be reported within 12 months after a trial’s PCD, but may be delayed until 30 days after FDA approval if a company files a ‘certificate of delay’ with the NIH.8 The other is that trial results are not due until 30 days after FDA approval of a new drug for an initial use approval. The Final Rule, a regulation released in September 2016 by the NIH and HHS, clarifies that trial results must be reported for both approved and unapproved indications, however, its effective date postdates the trials in our sample.21 We examined compliance with FDAAA among applicable trials using both interpretations and used the ‘approval date’ interpretation in calculating company rankings.
Lastly, we ranked NDA sponsors by their overall clinical trial transparency. For pharmaceutical companies with only one drug approved by the FDA in 2014, we averaged their scores on (1) the trials-in-patients analysis, excluding trials in healthy volunteers, and (2) a FDAAA compliance standard that counted a trial as compliant if it satisfied either interpretation of the reporting requirements. For companies with multiple drugs approved, we pooled the trials from all drugs and then calculated the percentages of trials satisfying each of the two standards. In rare cases, we excluded trials (n=12) from a particular company ranking if the NDA sponsor was not the trial sponsor and therefore not responsible for publicly reporting trial results. If the responsible party was a ranked company, and we could confirm they were indeed the responsible party, we transferred the trials to their denominator and included them in their rankings.
Statistical analysis
Summary statistics (medians and interquartile ranges) were calculated to show how commonly trials for each approved drug and drug company met the transparency measures. All data were collected and analysed in Microsoft Excel V.15.18 (Redmond, Washington, USA).
Validation
Datasets and results were sent to the NDA holders of each drug for validation (details in the online supplementary appendix 4). Nine of 11 evaluated companies participated in both the validation process and a meeting to discuss study methods and findings (details in the online supplementary appendix 5), affording validation for 79% of drugs reviewed. Validated results are presented below.
Results
In 2014, the FDA approved 31 new molecular entities (NMEs) or new combination drugs with at least 1 NME, 19 of which were sponsored by 11 of the 20 largest pharmaceutical or biotechnology companies (figure 1). A total of 553 trials (median of 24 trials per drug) were included in the NDAs.

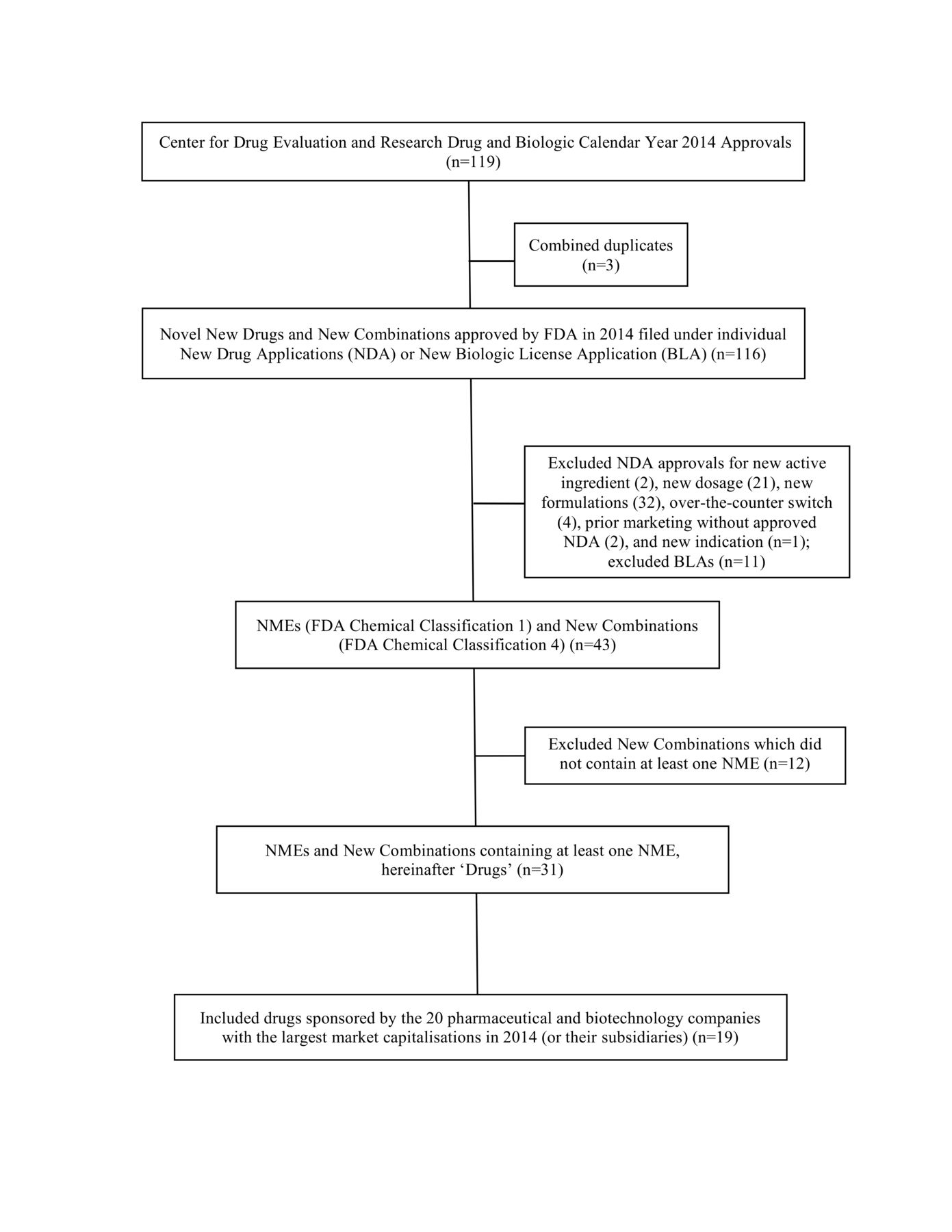{kind=link}
Drugs included in transparency analysis. BLA, biologic license application; FDA, Food and Drug Administration; NDA, new drug application; NME, new molecular entities.
We analysed 505 of these trials (median of 22 trials per drug), after excluding trials that were not at least 1 year past their PCD by our study cut-off of 1 February 2016, trials terminated without enrolment and expanded-access trials. Trials with unknown phases (n=7) were excluded from the FDAAA subsample. A median of seven trials per drug were conducted in patients. Trials in patients accounted for 233 of 505 trials, but 93% of all trial participants (124 664/133 428).
Transparency scores based on trials in patients
We first report results for the sample of trials in patients. A median of 100% (IQR 86%–100%) of trials in patients per drug were registered (table 2). A median of 71% (IQR 57%–100%) reported results or provided a CSR synopsis, and 80% (IQR 70%–100%) were published. Overall, results for a median of 96% (IQR 80%–100%) of trials, per drug, were publicly available in some form.
Transparency of clinical trials in patients and all clinical trials listed in NDAs
Ten of 19 drugs (53%) had at least one undisclosed trial conducted in patients. Six drugs (32%) had at least one undisclosed phase II or III trial. At least 2864 patients participated in trials with undisclosed results.
Transparency scores based on all trials in an NDA, including healthy volunteers
Transparency was lower using the standard that all trials in a successful NDA should be publicly available, than it was for the trials-in-patients standard (table 2). A median of 53% (IQR 33%–85%) of all trials per drug were registered, 24% (IQR 19%–50%) reported results or shared a CSR synopsis and 60% (IQR 37%–75%) were published. Overall, a median of 68% (IQR 40%–84%) of trial results per drug were publicly available (reported, shared in a CSR or published).
All 19 drugs had at least one publicly unavailable trial conducted in patients or healthy volunteers. Most of these trials were phase I trials involving healthy volunteers (median of 100% of trials, IQR 72%–100%). At least 7287 patients and healthy volunteers participated in trials with undisclosed results.
Compliance with FDAAA requirements
A median of 25% (IQR 19%–38%) of trials per drug (or four trials per drug) were subject to mandatory disclosures under FDAAA. Applying first the ‘trial completion date’ interpretation of FDAAA’s requirements, a median of 71% (IQR 0%–87%) of these trials per drug were FDAAA compliant (table 3). A median of 100% (IQR 100%–100%) of these trials per drug were registered on time and 71% (IQR 0%–87%) reported results on time. Of the 110 426 participants in trials covered by FDAAA, 66% were in non-compliant trials under this interpretation.
Compliance with FDAAA legal disclosure requirements
Applying the ‘approval date’ interpretation of the law, a median of 100% (IQR 75%–100%) of trials per drug were FDAAA compliant (table 3). A median of 100% (IQR 100%–100%) were registered on time and 100% (IQR 75%–100%) had results reported on time. A median of 100% (IQR 88%–100%) of participants were in compliant trials.
Most companies (73%) filed at least one certificate of delay for each of their drugs. However, a median of only 28% of FDAAA-applicable trials per drug had certificates of delay filed.
Timing of results reporting
At the time of FDA approval, results for a median of 45% (IQR 20%–52%) of all trials per drug are publicly available in some form (table 4). At 3 months postapproval, 50% (IQR 28%–67%) of all trials were publicly available. This median did not increase at 6 months postapproval, remaining at 50% (IQR 33%–73%).
Timing of public availability of trial results for new drugs approved by the FDA in 2014
For trials in patients, transparency was achieved more quickly. Results for a median of 65% (IQR 50%–73%) of trials were publicly available at the time of FDA approval. At 3 months postapproval, the median was 85% (IQR 67%–100%), and 86% (IQR 67%–100%) at 6 months.
Location of registration, reporting and CSRs
Almost all registered trials (315 of 316) were registered in ClinicalTrials.gov. No trials were uniquely registered or had summary results reported in international registries. Only one trial was uniquely registered in a corporate registry and not in ClinicalTrials.gov, no summary results were uniquely posted in corporate registries. While ClinicalTrials.gov contained occasional links to CSRs, it was not comprehensive; 41 CSRs were uniquely posted on corporate registries.
Company rankings
Sanofi/Genzyme and Johnson & Johnson/Janssen achieved the highest overall clinical trial transparency scores, tying for first place in the rankings and scoring 100% on the patient and FDAAA trial standards (table 5). AbbVie (96%), Celgene (95%), Merck (93%) and AstraZeneca also scored at or above the industry median. Valeant scored lowest (50%).
Companies’ overall clinical trial transparency rankings for drugs approved in 2014*
Discussion
This analysis of all known sources of publicly available information about clinical trials found high levels of transparency among large pharmaceutical companies and newly approved drugs for trials conducted in patients. Per drug, a median of 96% of trials in patients were publicly available in some form, 100% were registered, 71% reported results or shared CSRs and 80% were published, by 13 months after FDA approval. It takes about a year after FDA approval for companies to publicise most (96%) of their trials in patients. At FDA approval, 65% of trials per drug are available and at 3 months after FDA approval, 80%. Compliance with FDAAA requirements was high (median of 100% per drug).
The gap between transparency of results from the all-trials and trials-in-patients samples is striking. The median proportion of trials available per drug was markedly lower in the all-trials sample for trial registration (53% vs 100%), reporting results or CSR summaries (24% vs 71%), publication (60% vs 80%) and overall availability (68% vs 96%). The insomnia drug, Belsomra, is an extreme example of the difference: only 6 of 37 trials supporting the NDA were in patients, and the percentage publicly available was 100% for trials in patients but 20% among all trials (table 2).
Our earlier work examined transparency levels on these measures for trials in a successful NDA for drugs approved in 2012.25 Juxtaposing the two studies’ findings shows improved transparency levels on some standards. The median proportion of trials conducted in patients with publicly available results went up, from 87% in 2012 to 96% in 2014. Additionally, the proportion of drugs with undisclosed phase II or III trials went down, 50% vs 32%. However, there was little change in transparency levels on the all trials metrics (including trials in healthy volunteers). The median proportion of all trials registered per drug was 57% in 2012 vs 53% in 2014; the median for reporting results, 20% vs 24%; the median for publication, 56% vs 60%; and the median for overall availability, 65% vs 68%. The lack of increase in the proportions of trials registered and with results reported, on the all trials level, is surprising because our 2014 methodology newly incorporated more registries and included CSR synopses as satisfying the requirement for posting results. Compliance with FDAAA, measured by the ‘trial completion date’ interpretation, increased from a median of 67% of covered trials per drug in 2012 to 71% in 2014.
There is disagreement about the value of disclosing information for trials in healthy volunteers. Some pharmaceutical companies, pharmaceutical trade associations15 and commentators31 32 have focused on trials in patients for several reasons. Historically, later-phase efficacy trials have been considered to have the greatest public health relevance and salience for prescription guidelines writers and drug formulary committees. Limiting disclosure to trials in patients captures phase I trials for serious diseases, like most cancers, where the relevance of early data to patient care is high. Arguably, any important safety signals that emerge in phase I trials in healthy volunteers resurface in phase II trials, so critical safety information likely does reach the public. Lastly, trials in healthy volunteers are small, seldom controlled and therefore not generally powered to provide statistical significance.
Notwithstanding these arguments, there is value in making information about all trials available. NIH policy now requires it. Many companies already operate on this standard. It would alleviate public concerns about whether useful information is being hidden, and speed decision making based on safety signals. Additionally, disclosing phase I trials may help speed innovation and save money, particularly for small biotechnology companies, by preventing others from travelling down known dead-end pathways or empowering them to design better trials based on the lessons learnt from previous studies.
Whether transparency analyses focus on all trials or trials in patients, there is a need for clearer, more harmonised standards so that progress over time can be gauged and companies receive a consistent message about what is important for them to do. At a minimum, FDA should clarify which of the two interpretations of FDAAA was correct. The Final Rule is helpful in this regard in that it newly requires the reporting of trial results for both approved and unapproved indications.
More broadly, we believe the measures articulated in this study are useful, broadly acceptable and demonstrably workable to implement. In ongoing work, we will supplement them with a measure for patient-level data sharing—the new frontier in clinical trial transparency.
Our study has limitations. First, our decision not to count abstracts submitted to scientific conferences as ‘publications’ may be controversial. Second, some studies may be published after our study cut-off of 13 months post-FDA approval of a drug. Third, we attributed transparency scores, on the drug level, generally to the company that submitted a drug’s NDA although a few trials in the NDA were sponsored by other companies—typically, a company the NDA sponsor acquired (see the online supplementary appendix 7). NDA sponsors presumably had access to data from those trials in order to file the NDA that included them; however, one company reported being unable to edit the ClinicalTrials.gov entry created by the acquired company. This limitation only applied to the drug evaluations, not the company rankings, as these trials were often excluded from the company rankings. Also, it is worth noting that some trials in an NDA are for different indications than the approved indication. The FDA generally evaluates these trials as safety trials for the approved indication. Our analysis was limited to large companies, to drugs approved by the FDA in 2014 and to trials included in the relevant NDAs approved in 2014. Finally, our company rankings are not adjusted for the volume of trials conducted. Some may object that this disadvantages companies with a large number of trials, for whom compliance may be more resource intensive.
Conclusion
Our study shows that clinical trial transparency practices vary according to the standard used to measure them. On the drug level, about half of FDA-approved drugs have publicly disclosed results for all trials in patients who were included in our sample. On the company level, about 18% of large companies fully disclosed all such results and complied with FDAAA disclosure requirements. Per drug, among trials in patients, a median of 100% of trials were registered and 96% had publicly available trial results, in some form. Among large pharmaceutical companies, clinical trial transparency is highl based on many measures, although opportunities for improvement remain.
Momentum for greater clinical trial transparency will grow as we continue to experience its benefits. Legal requirements in FDAAA and NIH rules push the effort forward, along with efforts by other organisations like the Gates Foundation, WHO and Wellcome Trust, but reaching consensus on standards and monitoring and publicising companies’ adherence to emerging standards are also critical. Celebrating progress—and identifying where it is not occurring as quickly as it could—can move the field forward toward a shared vision of transparency and what it can achieve.
Acknowledgments
The authors thank the Laura and John Arnold Foundation; pharmaceutical company representatives who validated their datasets; and Yael Bree (Baruch student, NYU and BEI intern), Tamara Hardoby (NYU graduate and BEI researcher), Mindy Kresch (Baruch student, NYU and BEI intern), Rosa Macrito (Columbia Student, BEI researcher), and Luke Sleiter (NYU graduate, NYU Researcher, BEI researcher) for research assistance. All errors and conclusions are those of the authors only.
References
Footnotes
Contributors JM: conceived the study, supervised the collection and coding of data, led the data analysis and wrote the initial manuscript draft. NR: contributed to data collection, coding, and analysis. MMM, JSR and MW: provided guidance on the analytical approach, contributed to the interpretation of results, and drafted and revised the manuscript for critical intellectual content.
Funding This work was funded by a grant from the Laura and John Arnold Foundation. JSR: receives support through Yale University from Johnson and Johnson to develop methods of clinical trial data sharing, from Medtronic, Inc and the Food and Drug Administration (FDA) to develop methods for postmarket surveillance of medical devices, from the FDA to establish the Yale-Mayo Center for Excellence in Regulatory Science and Innovation (CERSI), from the Blue Cross Blue Shield Association to better understand medical technology evaluation, from the Centers of Medicare and Medicaid Services (CMS) to develop and maintain performance measures that are used for public reporting, and from the Laura and John Arnold Foundation to support the Collaboration on Research Integrity and Transparency (CRIT) at Yale.
Disclaimer The views expressed in the report are the authors’ and do not necessarily reflect those of the foundation.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Study data will be posted at bioethicsinternational.org upon publication of this article and can also be obtained from the lead author.