Article Text

Abstract
Introduction Responsible conduct of research implies that results of clinical trials should be completely and adequately reported. This article describes the design of a cohort study that aims to investigate the occurrence and the determinants of selective reporting in an inception cohort of all clinical drug trials that were reviewed by the Dutch Institutional Review Boards (IRBs) in 2007. It also describes the characteristics of the study cohort.
Methods and analysis In 2007, Dutch IRBs reviewed 622 clinical drug trials. For each trial, we assessed the stages of progress. We discriminated five intermediate stages and five definite stages. Intermediate stages of progress are: approved by an IRB; started inclusion; completed as planned; terminated early; published as article. The definite stages of progress are: rejected by an IRB; never started inclusion; not published as article; completely reported; selectively reported. We will use univariate and multivariate Cox regression models to identify trial characteristics associated with non-publication. We will identify seven trial-specific discrepancy items, including the objectives, inclusion and exclusion criteria, end points, sample size, additional analyses, type of population analysis and sponsor acknowledgement. The percentage of trials with discrepancies between the protocol and the publication will be scored. We will investigate the association between trial characteristics and the occurrence of discrepancies.
Ethics and dissemination No IRB-approval is required for this study. Access to confidential research protocols was provided by the Central Committee on Research Involving Human Subjects. We plan to finish data collection in June 2015, and expect to complete data cleaning, analysis and manuscript preparation within the next 3 months. Hence, a first draft of an article containing the results is expected before the end of October 2015.
- Selective reporting
- Non-publication
- Selective publication
- Responsible conduct of research
- Publication bias
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
- Selective reporting
- Non-publication
- Selective publication
- Responsible conduct of research
- Publication bias
Strengths and limitations of this study
The major strength of our study is that we investigate the occurrence of both the non-publication rate and selective publication in the same cohort. By addressing both issues, our analyses will likely offer more insight than most of the previous publications on this topic.
We use original protocol data, which enables us to assess discrepancies more completely and objectively than if we would have used trial registry data only. We will not have to depend on voluntary provision of access to the original protocols in our assessment of selective reporting, which is an important limitation of most other studies.
The most important limitation of our study is that we have to rely on the response to the questionnaire of the investigators and sponsors for verification of whether the study was published. Hence, non-response may introduce bias in our study. To assess the potential impact of non-response bias, we will compare characteristics between responders and non-responders.
Introduction
Responsible conduct of clinical research implies that results of clinical trials should be completely and adequately reported.1 ,2 However, a significant part of clinical trial results is never reported: on average, only 50% of clinical trials that are started are published in the scientific literature.3–20 As published reporting gives the nature or direction of the trial conclusions, incomplete reporting may result in publication bias.8 ,9 ,19 ,21–24 For example, if negative findings are more often not published than positive findings, the overall evidence synthesis will be biased, which can harm patients.25–27
Publishing negative results is sometimes judged irrelevant or uninteresting by the investigator, the journal editor or the sponsor of the trial.28 Negative trials, however, add valuable information to the body of evidence on the effects of the interventions studied. Moreover, publishing negative findings can prevent the start of unnecessary new clinical trials. This may make the use of resources for investigators and sponsors more efficient.29 ,30
Selective reporting of trial results comes in two forms. First, selective reporting can mean that the trial at issue is never published in the scientific literature (non-publication). This can be judged by searching for publications on trials included in an inception cohort, for example, using information from a trial register.6 ,12 ,16 ,31 Second, selective reporting may indicate that a trial is published in the scientific literature with changes, additions or omissions of study aspects or findings (selective publication).32–34 The second form of selective reporting is more subtle and can only be judged by comparing published reports to the full original study protocol.
Non-publication rates of 10–88% have been reported in the literature.3 ,5 ,7–12 ,14–19 Selective publication was identified by studying discrepancies between the protocol and publication in reporting end points, sample size, statistical methods and subgroup analysis.33 ,35–37
That non-publication and selective publication can lead to patient harm was also shown in clinical trials with drugs intended for marketing authorisation.15 ,38 ,39 Some new drugs had to be withdrawn from the market after additional data was revealed showing harmful effects. For example, clinical data on the new anti-inflammatory drug rofecoxib were neither published in the literature, nor revealed to the regulators.39 Other examples of non-publication and selective publication potentially resulting in patient harm include the antihypertensive drug reboxetine,38 and the antiarrhythmic drug lorcainide.22 The negative media attention about these and other drug trials has caused a decrease of the public's trust in the pharmaceutical industry and medical research.40 ,41 This had led to various codes and guidelines aiming at reducing selective reporting42–44 to be developed. However, recent research showed that these guidelines have only reduced selective reporting marginally.45 ,46
Most studies that investigate selective reporting use data from a public registry, like clinicaltrials.gov. However, not all clinical trials are registered in public registries, and details of the original trial protocol are often unclear or lacking because these registers often do not include full study protocols. Also, information published in public registries may be subject to selective reporting as well. The availability of the full and original trial protocol submitted to an Institutional Review Board (IRB) enables the tracking of the stages of progress of a study from the start. Therefore, to our opinion, starting with a series of consecutive full trial protocols submitted to an IRB in a defined time window and in a defined area is the best approach to examine non-publication and selective publication. To date, few studies have been carried out using this approach.47
We report the design of a study that aims to evaluate reporting practices in an inception cohort of clinical drug trials in the Netherlands. The primary objective of the study is to investigate non-publication and selective publication in an inception cohort of clinical drug trials. With regard to non-publication, we will identify factors associated with non-publication. With regard to selective publication, we will evaluate factors associated with discrepancies between the protocol and the publications of the trials. The secondary objective of this study is to investigate whether selective publication is associated with the direction of trial conclusions. Furthermore, we describe the characteristics of the study cohort.
Methods and analysis
Characteristics and data sources
We identified all clinical drug trials reviewed by the Dutch accredited IRBs48 between 1 January 2007 and 31 December 2007 (n=622). These trials define the inception cohort. According to previous studies, a 7-year time window is sufficient for most trials to recruit participants, collect data, prepare a manuscript and publish the manuscript.5 ,8 ,16
Also, we identified the characteristics of these trials (see online supplementary table S1). The used source was the General Assessment and Registration (GAR) form. This is a standard obligatory form that investigators submit to the IRB. For 194 trials, multiple therapeutic areas were indicated. Two investigators (CAvdB and CTMB) independently examined whether these trials could be reclassified to a single therapeutic area and reclassified the combination trials as one therapeutic area. Differences were solved by consensus after involving a third investigator (GHK). To reduce the large number of different therapeutic areas, we reclassified the variable to the International Classification of Diseases, V.10.49 This reclassification retained 11 therapeutic areas and 1 ‘other’ category.
From the trials included, we will extract data on the stages of progress, non-publication and selective publication. In addition to the public data sources and original trial protocols, we plan to send out a questionnaire to the investigators. An overview of the variables we plan to extract is presented in the data extraction form (see online supplementary table S2).
Stages of progress
For the 622 trials in the inception cohort, we will determine the various stages of progress (figure 1). For each clinical drug trial, we will discriminate 10 stages of progress. Of these, five are intermediate (meaning that further action is observed or possible), and five are definite (meaning that no further action is observed or possible). We named the stages of progress according to the flow of the cohort, shown in figure 1. The intermediate stages of progress are: B1 approved by IRB; C1 started inclusion; D1 completed as planned; D2 terminated early; E1 published as article. The definite stages of progress are: B2 rejected by IRB; C2 Never started inclusion; E2 Not published as article; F1 completely reported; F2 selectively reported. We primarily aim to investigate the publication-related stages of progress E1, E2, F1 and F2. However, to understand why these stages of progress are not reached, we also determine the other stages of progress. The stage of progress F2 (selectively reported) is definite for the end of our data collection; later publications can still fill the remaining gaps by moving trials to F1 (completely reported).

Overview of stages of progress of the 2007 inception cohort of clinical drug trials in the Netherlands. The numbers in the boxes indicate the numbers of trials that succeeded to the specific stages of progress. From B1, C1, D1, E1 to F1 is the ‘perfect’ flow of a trial in the cohort, meaning that all aspects took place according to the application. The sum of the boxes B2, C2, E2 F1 and F2, which are the five final stages of progress, will be 622.
Non-publication
We search for publications on the trial results in the scientific literature using a standardised algorithm (figure 2). A publication is defined as a peer-reviewed article containing at least methods and results. All reports not fulfilling this publication (eg, results reported in registries, conference abstracts containing results, trial summaries on sponsor websites containing methods and results) will be also collected. Peer-reviewed publication is in our opinion the golden standard for reporting clinical research, but trial results can be reported by other means (eg, registries, sponsor websites, conference abstracts). Using only peer-reviewed articles as end point for non-publication is in line with the majority of other research.47 If we identify more than one publication of trial results, we classify the publication as either primary (ie, containing the overall results and conclusions) or secondary (ie, interim, post hoc, subgroup or other analysis). In general, we assume that this will be clearly stated in the publications.42 Other information collected includes the full-text of the article, the journal and the first date of publication (eg, advance online publication). We have completed this part of the publication search in March 2015.

{kind=link}
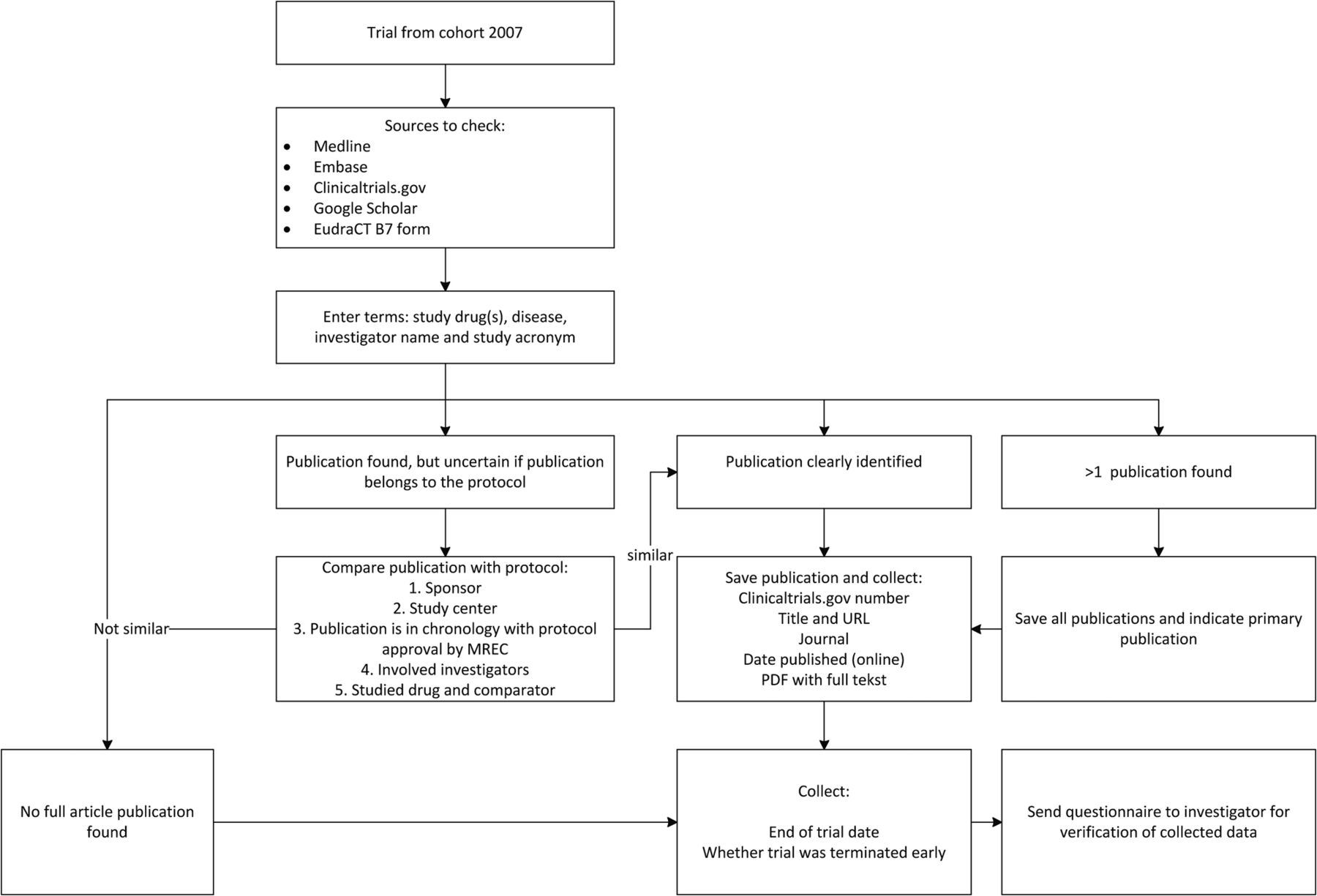{kind=link}
Publication search algorithm. EudraCT, European Union Drug Regulating Authorities Clinical Trials: obligatory registration database for clinical drug trials carried out in the European Union.
We will also collect the end of trial date and information about (early) termination of the trial. We define the end of trial date as the date of the last visit of the last patient undergoing the trial.50 A trial is terminated early if either the inclusion or the follow-up is terminated earlier than foreseen in the research protocol. Since early termination is an intermediate stage of progress of a trial, we include early termination as a potential determinant for the end points studied. In addition, prospective registration on clinicaltrials.gov will be examined as a potential determinant (table 1). We define prospective registration as registration of the trial before the first patient is recruited.1 The data field ‘first received’ on clinicaltrials.gov will be used as the date of registration.
Planned analyses, end points and determinants
To validate the used publication search algorithm, two investigators independently searched for publications using the algorithm, using a random selection of 30 trials of the cohort. The two searches identified no differences. We checked the external validity of the algorithm by comparing the results to a search algorithm used for another study,33 provided by the investigators. This comparison showed no differences, which suggested that the construct validity of our algorithm was adequate.
In addition, we will send questionnaires to the main investigators of the research divisions or hospital departments that conducted the trials. We will specifically ask the investigators to confirm or rectify our information about which stage of progress the trial reached according to our findings. For the non-published trials, we ask for the reasons that the trial was not published (box 1) and whether the results of the trial were reported in alternative ways, such as on clinicaltrials.gov. When the investigator does not respond to the mailed questionnaire, we will try to engage the investigator by telephone contact. In case we are unable to contact the investigator, we will contact the sponsor of the trial.
Reasons for not publishing results, to be obtained from the questionnaire (for unpublished completed trials in cohort)
▸ Manuscript is in preparation/under review
▸ Results were not interesting enough to publish
▸ Journal rejected the manuscript
▸ Sponsor decided not to publish without providing a reason
▸ Other
The various stages of progress of the trial in the flowchart will be updated according to the results of the questionnaire. In case neither the investigator nor the sponsor could be reached, the stages of progress remain unchanged. We assume that if a trial was incorrectly placed in the stage of progress boxes C2, D2, or E2, the investigator or sponsor would have responded. If we are unable to find any information on whether a trial started inclusion, ended or was published, we will exclude the trial for subsequent analysis. After showing construct validity, the publication search was performed by two authors (CAvdB and MvH), double-checked by the questionnaire to the investigators. To assess the likelihood of bias, we will investigate whether the characteristics of included cases differ from excluded cases.
Selective publication
Among the trial protocols that resulted in a publication, we will further investigate selective publication. We include only peer-reviewed articles for the discrepancy analysis because other reports contain too little detail to investigate discrepancies with the trial protocol. Selective publication can be measured by identifying discrepancies between protocol and publication. Discrepancies between protocol and publication are indications of selective publication, which may lead to reporting bias. The degree of the risk of reporting bias depends on the association of discrepancies with the direction of trial conclusions. Therefore, among the trials with a randomised design, we will also assess the direction of publication conclusions and investigate whether the direction of publication conclusions is associated with discrepancies between protocol and publication.
We define discrepancies between protocol and primary publication as additions, omissions or changes in prespecified discrepancy-items. To identify discrepancies systematically, we developed an extraction form containing relevant items. We used items from common protocol and publication guidelines like SPIRIT and CONSORT to compel a list with trial items that should be reported. From that list, we selected seven items in which we expected selective reporting (see online supplementary table S2).33 ,36 ,37 The seven discrepancy items include: (1) objectives, (2) inclusion and exclusion criteria, (3) end points, (4) sample size, (5) additional analyses, (6) type of population analysis and (7) sponsor acknowledgement. We will extract these items both from the protocols and the publications. Subsequently, we will compare the extracted data of the protocol to the publications. With regard to discrepancies in the objectives and end points, we will distinguish between discrepancies in the primary and in the secondary objectives, and end points. With regard to discrepancies in the inclusion and exclusion criteria, we will only consider an objective change as discrepancy because inclusion and exclusion criteria are often not fully reported in publications due to the limited availability of space. We will operationalise discrepancies in the planned versus included sample size as the ratio of sample size achieved divided by sample size planned. With regard to discrepancies in the type of population analysis, we will assess whether an intention-to-treat or per-protocol analysis was planned and used accordingly. We will also indicate when there was a lack of information in the protocol and/or in the publication to assess a discrepancy.
In case we identify multiple publications of one trial protocol, we will include the primary publication in the discrepancy assessment. In addition, if a secondary publication contains any analyses that were not described in the study protocol and this was not stated in the publication, we classify that as an additional discrepancy.
The discrepancy assessment was developed by one author (CAvdB), and will be tested for construct validity by a second author (PCS) by performing an independent discrepancy assessment of a random selection of 10% of the published trials. Remaining differences will be solved by discussing these with two other authors (CTMB and SWJJ). The remaining trials will then be assessed by one author (CAvdB), with a randomly selected double-check of 20 of the published trials by a second author (PCS). Uncertainties will be solved by a discussion involving two other authors (CTMB and SWJJ).
Among the trials with a randomised design, we will classify the direction of publication conclusions as either positive or negative. This classification is included to investigate whether discrepancies are associated with the direction of the conclusions (and the interpretation) that the authors draw in the discussion sections of the publications. If trials with a positive conclusion have more discrepancies than trials with a negative conclusion, this may mean that discrepancies are used to spin trial conclusions towards a positive direction. Two independent investigators (CAvdB and PCS) will independently classify the trials, and solve differences by consensus.
Data analysis
According to the objectives of the study, we will analyse three end points (table 1): non-publication, discrepancies between the protocol and the publication as a proxy for selective publication and the direction of publication conclusions.
Non-publication
In a survival analysis of the non-publication rate, only trials that started inclusion will be analysed (box C1 of figure 1). The end point used is non-publication as peer-reviewed article, according to the definition provided above. The trial end date marks the start of follow-up (ie, the date the trial transits to the stage of progress D1 or D2, figure 1). We chose this date instead of the date of IRB approval because the trials in the cohort might differ in time span. This time span may depend, for example, on the phase of the trial and the number of participants to be recruited. In case of multiple publications of one trial protocol, we use the publication date of the primary publication.
We assume that all trials that started including patients are eligible for publication. Thus, the population of the non-publication survival analysis includes all trials that started inclusion (box C1, figure 1). Trials that never started inclusion are excluded from this analysis.
To identify characteristics that are associated with (non-)publication, we perform Cox regression analysis to estimate the strength of the association between characteristics and publication status, expressed as HRs and 95% CIs. Since trials of oncolytic drugs are different with respect to the disease severity compared to most trials in other therapeutic areas (which may affect publication), a stratified analysis will be conducted as well. In addition, we will tabulate reasons for non-publication. Finally, we will describe the means of publication by other means than by the definition of publication. By doing so, we will identify the subset of trials with no results reported at all (not as peer-reviewed article and not by any other means).
Selective publication
For each of the seven discrepancy-items, we calculate the proportion of trials with the discrepancy. We investigate the association between characteristics and discrepancies for each item (χ2 test) and for the total discrepancy summary score (paired t test). We will use multivariate logistic (individual discrepancies) and linear (total discrepancy score) regression models to estimate the strength of the association of characteristics and publication status, expressed as ORs and 95% CIs. Among the trials with a randomised design, we investigate whether the discrepancies are associated with the direction of the publication conclusions using identical bivariate and multivariate analyses. Data analysis will be performed by two authors (CAvdB and PCS), and double-checked by all other authors.
By measuring non-publication and selective publication, the study will identify the extent of research under-reporting waste in a cohort of clinical trials in the Netherlands.51 ,52 To increase the value derived from clinical trials, transparency from protocol to the public is needed.53 Our study will provide this on a national level and may elucidate areas for improvement. Ultimately, this study may contribute to evidence-based medicine by improving the unbiased reporting rates of clinical drug trials. This may increase the overall trust in research on drugs and the willingness of participants to enrol in clinical drug trials.
Ethics and dissemination
As our study involves no human participants, no IRB-approval is required. Access to confidential research protocols was provided by the Central Committee on Research Involving Human Subjects. We plan to finish data collection in June 2015, and expect to complete data cleaning, analysis and manuscript preparation within the next 3 months. Hence, a first draft of an article containing the results is expected before the end of October 2015.
Acknowledgments
The authors thank our colleagues from the Central Committee on Research Involving Human Subjects (competent authority governing all accredited IRBs in the Netherlands) for their collaboration. In particular, we acknowledge Dr Monique Al and Ms Miranda Vermeulen for their assistance in the data extraction procedure.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
Footnotes
Collaborators Central Committee on Research involving Human Subjects.
Contributors All authors except MvH engaged from the start of the project in discussing and structuring the methodology. MvH is involved as a visiting scientist in data collection and analysis from August 2014 until January 2015. CAvdB, PCS, CTMB and GHK were involved in analysing the baseline trial characteristics of the cohort. CAvdB was as first author responsible for drafting the manuscript. All other authors provided feedback on the full text, on all tables and figures, and on the abstract.
Funding Ministry of Health, Welfare and Sports (unrestricted grant).
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Original study protocols and individual study characteristics were accessible to CAvdB, PCS, MvH and CTMB through the archives of the Central Committee on Research involving Human Subjects. Full texts of publications were downloaded and accessed through inst.