Article Text

Abstract
Objective Aerobic exercise can improve cardiovascular fitness and does not seem to be detrimental to patients with asthma, though its role in changing asthma control and inflammatory profiles is unclear. The main hypothesis of the current randomised controlled trial is that aerobic exercise will be superior to usual care in improving asthma control. Key secondary outcomes are asthma quality of life and inflammatory profiles.
Design A total of 104 sedentary adults with physician-diagnosed asthma will be recruited. Eligible participants will undergo a series of baseline assessments including: the asthma control questionnaire; the asthma quality-of-life questionnaire and the inflammatory profile (assessed from both the blood and sputum samples). On completion of the assessments, participants will be randomised (1:1 allocation) to either 12-weeks of usual care or usual care plus aerobic exercise. Aerobic exercise will consist of three supervised training sessions per week. Each session will consist of taking a short-acting bronchodilator, 10 min of warm-up, 40 min of aerobic exercise (50–75% of heart rate reserve for weeks 1–4, then 70–85% for weeks 5–12) and a 10 min cool-down. Within 1 week of completion, participants will be reassessed (same battery as at baseline). Analyses will assess the difference between the two intervention arms on postintervention levels of asthma control, quality of life and inflammation, adjusting for age, baseline inhaled corticosteroid prescription, body weight change and pretreatment dependent variable level. Missing data will be handled using standard multiple imputation techniques.
Ethics and dissemination The study has been approved by all relevant research ethics boards. Written consent will be obtained from all participants who will be able to withdraw at any time.
Results The result will be disseminated to three groups of stakeholder groups: (1) the scientific and professional community; (2) the research participants and (3) the general public.
Registration Details ClinicalTrials.gov Identifier NCT00953342
- Exercise
- Quality of life
- Inflammation
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/3.0/ and http://creativecommons.org/licenses/by-nc/3.0/legalcode
Statistics from Altmetric.com
Introduction
Asthma is a chronic disorder of the airways characterised by reversible and intermittent airway obstruction and airway inflammation in response to a variety of stimuli (eg, pollen, dust, animal hair, smoke and airborne pollutants). Asthma is among the four most common chronic disorders affecting Canadian adults1 ,2 with over 2.5 million (8.5%) adults being diagnosed as having asthma.3 More importantly, asthma is an escalating medical problem in Canada, with a 40% increase in asthma incidence in the last two decades.3 ,4 Furthermore, the rates of poor asthma control have also increased, with nearly 60% of Canadian patients being poorly controlled.5 ,6 These increases have come despite important advances in diagnosis and treatment,7–11 suggesting that these changes in asthma must be multifactorial.12 Cytokines have been shown to play a critical role in orchestrating, perpetuating and amplifying the inflammatory response in asthma. Among these cytokines, interleukin (IL)-1, tumour necrosis factor (TNF)-α and IL-6 are involved in chronic airway diseases, while IL-4, IL-5, IL-9 and IL-13, which are mainly derived from T helper type (Th)-2 cells, are more specific to allergic inflammation.13 IL-4 is an upstream cytokine that regulates allergic inflammation by promoting Th2 cell differentiation and immunoglobulin (Ig) G switching to IgE subsequently, whereas IL-5 is highly specific for inducing eosinophilic inflammation.13 Similar to IL-4, IL-13 regulates IgE production but, unlike IL-4, it does not regulate T cell differentiation to Th2 cells.14 Thus, the increased and abnormal expression of cytokines in airways is one of the major features of allergic asthma. Recent reports have indicated that stimulating Th1 cells (IL-2 and interferon (IFN)-γ) might suppress Th2 cells, and thereby allergic inflammation.15 Therefore, the balance between Th1 and Th2 is important for immunoregulation, with imbalance believed to cause allergic asthma, and may predict the chronicity and the severity of asthma. However, recent findings indicate that the overall picture is more complex. For instance, both endogenous and inducible CD4+CD25+ regulatory T cells, through the release of IL-10, inhibit inappropriate immune responses in Th1/Th2 balance.16
A recent Cochrane review on physical training (aerobic exercise intervention of at least 20 min twice a week for 4 weeks) and asthma (which included articles up to April 2011)17 reported the following key findings:
-
Training improved cardiopulmonary fitness as measured by a statistically and clinically significant increase in maximum oxygen uptake;
-
Training had no significant effect on resting lung function;
-
None of the studies reported a worsening of asthma symptoms following training;
-
Training may have a positive effect on health-related quality of life.
These findings lead the authors to conclude that “… training can improve cardiopulmonary fitness and was well tolerated among people with asthma” and that this can be performed ‘… without fear of symptom exacerbation.”17 However, it should be noted that only 19 studies (695 participants) were included in the review, and overall these studies were of low quality with a high risk of bias. As such, it would seem that aerobic exercise is safe, increases fitness, does not change lung function, and may improve quality of life. With regard to asthma symptoms, only one of the seven studies reporting on symptoms actually used a recognised questionnaire (the Asthma Control Questionnaire (ACQ)18) to assess symptoms,19 with no changes seen between the exercise or control interventions. However, a small randomised controlled trial, which was published after the Cochrane review, found that aerobic exercise was associated with a statistical and clinical improvement in control in patients with asthma. These findings suggest that still more work needs to be done to understand the effects of exercise in symptom management and asthma control. Finally, though not the focus of the Cochrane review, one of the included studies found that aerobic exercise had a positive impact on inflammation in patients with asthma. Using induced sputum cell counts, Mendes et al20 reported decreases in eosinophils in the exercise group compared with the control group. While this result suggests a benefit of aerobic exercise, the data are still too limited to come to any solid conclusions.
Originality and importance of the results
The therapeutic role of exercise training has been established for various chronic conditions (eg, heart disease, chronic obstructive pulmonary disease (COPD), type II diabetes, etc) such that this non-pharmacological intervention is now included in several disease management guidelines. To our knowledge, the proposed research project will be one of the first to systematically evaluate the benefits of aerobic exercise training on asthma control, and the potential for inflammation to underpin such benefits, in adult asthma patients. The results of this study will add to the current evidence base for physicians to prescribe aerobic exercise for patients with asthma.
Research hypothesis
Aerobic exercise will be superior to usual care in the treatment of asthma control and other measures of morbidity.
Study objectives
Primary objective
To determine if, in comparison to usual care, a 12-week supervised aerobic exercise programme improves asthma control in patients with stable physician-diagnosed asthma.
Secondary objectives
Key secondary objectives
To determine if, in comparison to usual care, a 12-week supervised aerobic exercise programme improves asthma quality of lifeE and inflammatory profiles in patients with stable physician-diagnosed asthma.
Other secondary objectives
To assess potential inflammatory mechanisms linking aerobic exercise and improved asthma control.
Methods and analysis
Trial design
The ex-asthma study trial is designed as a randomised, controlled, observer-blinded multicentre superiority trial with two parallel groups and a primary end point of asthma control (Juniper questionnaire) in the week following the intervention period. Randomisation will be performed with a 1:1 computer generated allocation.
Study setting
To ensure that an objective physician diagnosis of asthma, and to increase the probability of detecting a higher level of poor asthma control, patients will be primarily recruited from the outpatient asthma clinics at Hôpital du Sacré-Cœur de Montréal (HSCM), the Montreal Chest Institute and the Jewish General Hospital. All sites are based in Montreal to ensure ease of oversight in the delivery of the study.
Eligibility criteria
Patients must provide written informed consent before any study procedures are performed. To determine eligibility and the stability of the patients’ asthma control and medication, potential participants will undergo a 4-week screening period.
Inclusion criteria:
-
Physician-diagnosed asthma (confirmed by medical record evidence of bronchodilator reversibility of 12% or PC20 methacholine ≤16 mg/mL);
-
Sedentary (currently do less than 60 min of structured/planned physical activity per week);
-
Taking at least 250 mg fluticasone equivalent per day;
-
On stable dose and regimen of asthma medications;
-
Mild-to-moderate symptomatic asthma as defined by an ACQ score of 1.25 or greater.21
Exclusion criteria:
-
Diagnosed comorbid disease for which there are already established exercise guidelines, that is, cardiac disease or COPD;
-
Any other medical condition that confers greater illness morbidity than asthma (eg, active cancer) which will be confirmed by physician review;
-
Forced expiratory volume in 1 s (FEV1) lower than 60% of predicted;
-
Incapable of exercising;
-
A body mass index >30 kg/m2;
-
Unable to speak or understand either French or English;
-
<18 years of age
-
Patients who are currently pregnant or intend to become pregnant over the course of the study.
Interventions
Aerobic exercise intervention
The exercise intervention will start within a week of completing the final baseline assessment. Participants will exercise three times/week at a level of 50–75% of their initial heart rate reserve determined at the time of the baseline exercise test for the first 4 weeks, and then progress to 70–85% of their heart rate reserve for the remaining 8 weeks.22 Patients will be required to take their reliever medication (short-acting bronchodilator) approximately 15 min prior to starting the exercise session (as per the GINA guidelines8) to reduce the chances of exercise-induced bronchospasm. The exercise routine will consist of 10 min of warm-up exercises, 40 min of biking and/or walking (and eventually jogging) and 10 min of cool-down exercises. Emphasis will be placed on ensuring that patients cool down properly as this is the time when most exercise-induced bronchospasm (EIB) events would potentially occur.23–26 A trained exercise physiologist (Canadian Sport and Exercise Physiologist certified or equivalent) will supervise all exercise sessions, and will perform three checks of heart rates per session to ensure that participants are exercising at a sufficient intensity. In general, this exercise programme follows the guidelines for patients with chronic illnesses (eg, patients with heart disease and COPD).27 ,28 In total, we offer six time slots per week (3 in the morning before work and 3 in the afternoon after work). All participants will be required to maintain their normal medical regimens during the intervention. Exercise sessions will be conducted at either the Centre de readaptation Jean-Jacques Gauthier, HSCM or within the pulmonary rehabilitation programme, Montreal Chest Institute.
Usual medical care comparison group
Patients in the usual medical care control comparison group will be asked to refrain from any structured exercise (ie, maintain their current behaviour) for 12 weeks until they are re-evaluated. During this 12-week period, we will contact patients every 4 weeks to assess asthma symptoms, events, medication adherence and check to see if they have started or intend to start exercising. Based on our previous studies, only about 1% of control participants actively engage in exercise.29 To ensure equity to all our patients, participants in the comparison group will be given the opportunity to participate in the exercise programme once they have completed the post-trial assessments. No assessments will be made following the cross-over exercise period.
Adherence to the exercise protocol
We will take the following steps to maximise adherence to the interventions: (1) Careful screening: we will thoroughly review our protocol and emphasise the importance of recruiting motivated participants who are ready to embark upon a sustained exercise programme. (2) Thorough orientation: participants will receive detailed information about the rationale and potential benefits of the programme. (3) Behavioural contracts: a written contract will be signed by the participant at the time of the initial orientation. In this contract, the patient will agree to participate in the programme to the best of their abilities, and to discuss any concerns or difficulties with the study coordinator before considering dropping out of the programme. (4) Close exercise supervision: during the exercise sessions, we will closely supervise exercising participants to ensure that they exercise within their prescribed training ranges. In addition, the participants’ heart rate will be consistently monitored so that patients who fall below or who exceed their respective training ranges can be identified and their exercise modified. This approach has been used successfully in our prior work.29 These strategies have been successful in our prior exercise studies employing patients with chronic diseases, where the dropout rates were <5%.29
Concomitant care
As both arms of the study will be receiving the usual medical care, all potential addition treatment options are available to the patient's regular physicians. However, if there are any additional medical interventions, they will be noted in the patient’s chart.
Outcomes
Primary outcome measure
Differences between the two treatment arms in the absolute change asthma control postintervention will be assessed using the ACQ.18 ACQ evaluates asthma control by asking respondents to recall their symptoms (including nocturnal awakenings), activity limitation and bronchodilator use in the last week. An additional question assessing FEV1, % predicted, is completed by the research assistant. It contains seven items rated on a seven-point scale (0=good control, 6=poor control). It has high intraclass correlation coefficients (0.90–0.95) and good construct, cross-sectional and longitudinal validity.18 ,30 ,31 It has also been shown that a score greater than 0.8 is associated with poor control,21 and a change or difference of ≥0.5 in ACQ has clinical significance.31 ACQ will be administered within 1 week of the completion of the intervention.
Secondary outcomes measures
In addition to ACQ, absolute differences in the Asthma Control Test (ACT)32 ,33 will be used as a secondary measure of asthma control. ACT evaluates levels of asthma control according to standard criteria specified by international guidelines:34 activity limitations, shortness of breath, nocturnal or waking symptoms, bronchodilator use and perceived control over the past 4 weeks. It contains five items rated on a five-point scale (with higher scores indicating better control). ACT has also demonstrated excellent measurement properties, including good internal consistency (α=0.79–0.85), test–retest reliability (α=0.77) and excellent criterion validity with ACQ (r=−0.89). A score of ≤19 on this questionnaire is indicative of poor asthma control.33 ACT will be administered within 1 week of the completion of the intervention.
Differences between the two treatment arms in the absolute change in asthma-related quality of life postintervention will be assessed by the Asthma Quality of Life Questionnaire (AQLQ).35 AQLQ assesses the extent to which asthma limits the asthmatic's life or interferes with their ability to do and/or enjoy different activities typical of daily life. AQLQ has 32 items rated on a seven-point Likert scale from 1 (a great deal) to 7 (not at all). It has four subscales: activity limitation, symptoms, environment and emotional distress. It has high intraclass correlation coefficients (0.90–0.95) and good construct, cross-sectional and longitudinal validity.35–37 It has also been shown that a change or difference of ≥0.5 in AQLQ has clinical significance.38 AQLQ will be administered within 1 week of the completion of the intervention.
Differences between the two treatment arms in inflammatory profiles postintervention will be assessed using a variety of sputum-based and blood-based markers. Sputum will be induced by the method described by Pin et al39 and modified by that of Pizzichini et al40 Briefly, participants will be pretreated with 200 μg of salbutamol before inhaling increasing concentrations of hypertonic saline (3%, 4% and 5%) for 7 min each (for a maximum of 21 min) with a Medix electronic nebuliser (Medix, Catthorp, England) without a valve or nose clip. After each inhalation, participants will be instructed to blow their nose, rinse their mouth and swallow the water to minimise postnasal drip and squamous epithelial cell contamination, respectively, before trying to expectorate in a sterile container. The mucus that will be obtained will then be separated from the saliva using forceps, weighed and rocked with four times its volume of dithiothreitol (Sputolysin; Calbiochem Corp., La Jolla, California, USA) for 15 min. The reaction will be stopped by adding an equal volume of Dulbecco’s phosphate-buffered saline (D-PBS) 1×(Invitrogen, Burlington, Ontario, Canada). The cellular suspension will then be centrifuged at 8000 rpm for 4 min and the supernatant will be collected and frozen at −80°C for further analysis. The cells will then be resuspended in D-PBS 1× and slides will be prepared with a Cytospin 3 (Shandon Scientific Ltd, Astmoor, England) and coloured with Diff-Quik solutions (Dade Diagnostics Inc, Aguada, Puerto Rico, USA) for a count of 400 cells. Sputum supernatants will be concentrated using Amicon columns before assessing the mediator’s quantification, as described previously.41 The sputum supernatant eosinophil cationic protein (a marker of eosinophils activation) and neutrophil-derived myeloperoxidase (a marker of neutrophils activation) will be determined by ELISA (BioSource, Medicorp).41 The Th2 cytokines, IL-4, IL-13, IL-10, the Th1 cytokines, IL-2, IFN-γ, IL-12 and the acute inflammatory cytokines IL-6 and TNF-α will be determined using a mutiplex bead array cytokine assay and Luminex technology.42 The Bio-Plex system (Bio-Rad) was recently acquired by our research group from a CIHR grant (# PRG 80172). Bio-Plex cytokine kits and complementary kits (eg, a Bio-Plex cytokine reagent kit) will allow the simultaneous quantification of several human cytokines in a single 10–25 mL biological sample with an extended dynamic range compared with ELISA. Cytokines will be measured according to the manufacturer's instructions. Using standard hospital procedures, blood draws will be made following a 20-min rest period via the insertion of an 18-gauge needle into the anterior cubital vein of the non-dominant arm. A 5 cc of blood will be drawn into tubes, and the serum will be collected and stored at, 80 C until assayed for cytokines levels (see details below). In addition, 5 cc of blood will be drawn into heparinised tubes for differential white cell counts, conducted on a Coulter STKS counter.
Other notable outcomes
As a series of manipulation checks and based on previous studies, we are anticipating that there will be a postintervention difference in exercise capacity between the two intervention groups but no difference in lung function.43 ,44
As per the guidelines,45 all participants will undergo standard fitness evaluations using an electrically braked cycle ergometer (Collins CPX Bike model 0070; Warren E Collins; Braintree, Massachusetts, USA) following the Jones protocol.46 Participants will exercise to exhaustion (fatigue, leg pain or shortness of breath). Expired air will be collected through the mouthpiece of a calibrated screen pneumotachograph (Collins/Cybermedic model 003500; Warren E Collins) and into an O2 and CO2 analyser (Quinton Qplex; A-H Robins, Bothel, Washington, USA). The pneumotachograph will be located 5 cm from the mouth and will be calibrated using the American Thoracic Society standards.47 As per standard practice,27 ,48 minute ventilation (VE), oxygen uptake (VO2) and CO2 excretion (VCO2) will be analysed on a breath-by-breath basis, with minute averages being obtained during the rest period (2 min of standing) and exercise test. To decrease the chances of patients having an EIB, all participants will be required to take their reliever medication 15 min prior to starting the exercise and will participate in active recovery/cool down.
Airway responsiveness to methacholine will be assessed according to the methods described previously.49 Aerosols will be generated by a Wright nebuliser at 344 Kpa and 8 L min in order to get an output of 0.13 mL min. The aerosol will be inhaled by tidal breathing, with a face-mask held loosely over the face and a noseclip. Half-doubling concentrations of methacholine (from 0.03 to 128 mg/mL) will be inhaled for 2 min at 5 min intervals, until a fall of at least 20% of the FEV1. Airway responsiveness will be expressed as the PC20 methacholine obtained from the log dose–response curve.
Standard pulmonary function tests, including spirometry, lung volumes and diffusion capacity, will be obtained according to previously described guidelines.47 ,50 For the assessments, rescue (bronchodilator) medication will be withheld for at least 4 h before pulmonary function tests are performed. FEV1 and forced vital capacity will be assessed before and 15 min after the administration of 200 μg salbutamol using a metered-dose inhaler or 500 µg terbutaline using a Turbuhaler. All results will be related to standard normal values.51–54
Additional measures
To be able to appropriately describe our population, and in parallel with our previous research,55–57 we will collect basic demographic and medical information, most notable of which will be age, which will be used for determination of eligibility. In addition, health behaviours (smoking status,58 diet habits59 and sleep60), ethnicity, marital status, socioeconomic status (years of education, income, occupation and residential deprivation61 ,62), gender role63 and anthropometrics (height, weight, and waist and hip circumference) will also be collected. The self-reported details of current and lifetime histories of asthma, other respiratory diseases, cardiovascular disease (CVD), CVD risk factors, cancers and auto-immune diseases, as well as all current medications will be collected. We will also administer the Morisky Self-Rated Measure of Medication Adherence questionnaire,64 which is a validated measure of medication adherence.65 It will be used to assess inhaled corticosteroid adherence. In addition, we will ask if patients are currently pregnant or intend to become pregnant over the course of the study for exclusion purposes. All reported clinical data will be verified by a hospital medical record review.
Participant timeline
The aerobic exercise intervention will start within 1 week of completion of the baseline assessments and the usual care intervention will commence on completion of the baseline assessments. The follow-up assessments will be the same as those completed at baseline and will occur within 1 week of the end of the intervention, that is, 13 weeks after the baseline assessments. However, to ensure there are no carry-over effects of acute exercise bouts, all postintervention measures will be assessed after a minimum of 24 h of inactivity (recovery).66 See figure 1 for a schematic of the study.
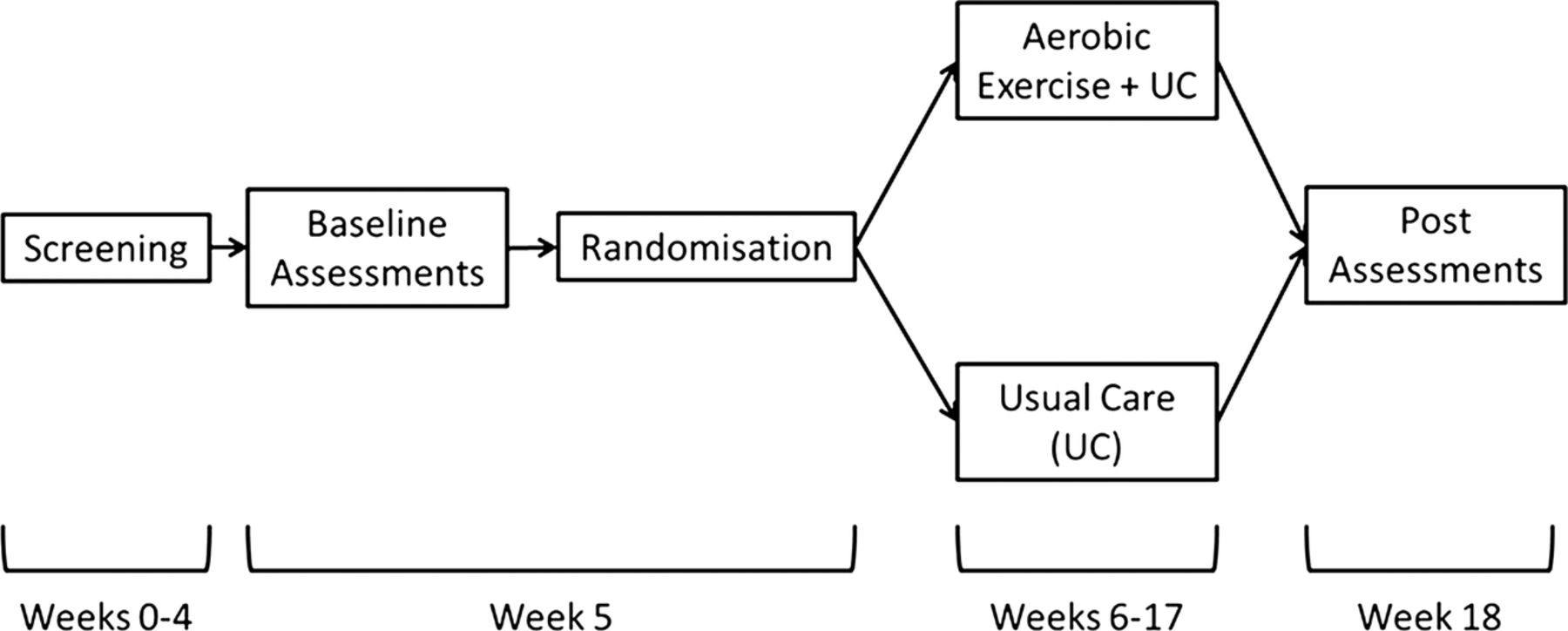
{kind=link}
Schematic of participant timeline.
Sample size
We have based our sample size calculations on data that we have already collected from participants who were recruited from the asthma clinic at HSCM. Using data from our current epidemiological sample55–57 and the inclusion/exclusion criteria detailed above, we established that for the current eligible group, the baseline mean±SD ACQ score would be 2.14±0.86. Using a standard 2 group design with equal variance and setting power at 0.8, p=0.05, and two-sided hypothesis testing, a decrease of 0.5 in the ACQ score (this change has been shown to be clinically significant31) would require a sample size of 47/group, for a total sample of 94. Using the same criteria, we established that the baseline mean±SD AQLQ score for the eligible group is 4.60±0.91, and using the same parameters, a clinically significant increase of 0.538 in AQLQ would require a sample size of 52 patients/group for a total sample size of 104. To deal with the issue of compliance and dropouts, as recommended by the CONSORT statement,67 we will use intent-to-treat analyses.68 Use of the intent-to-treat analyses preserves the effects of group allocation and provides an assessment of the practical impact of a treatment. As there are no data on the effects of exercise training on immune markers in asthma patients, and few clinical guidelines on which to base expected changes, we have used published data from a previous study in a diseased population. Like asthma, heart disease is also considered a disease of immune dysfunction; as such, using data generated by Goldhammer et al69 for an exercise training intervention on coronary heart disease patients seems to be an adequate estimation of the immune effects we may see in asthma patients. For example, setting a power of 0.8, and p=0.05, the required sample size to see a similar outcome in the current study for IL-1, IL-6 and INF-γ would be 34, 36 and 78, respectively. All these sample size estimates are within the proposed sample size of 104.
Recruitment
The primary recruitment site will be HSCM (ca. 70% of participants) with the other two sites each contributing about 15% of the total required participants. All three sites will use patient lists for those attending asthma or pneumology clinics to identify potentially eligible patients to approach; in general, each site sees over 20 asthma patients per week. In addition, the HSCM site will use a pre-existing database of over 800 tertiary care asthma patients,55–57 from which eligible patients will be contacted, and local advertising via newspapers and radio to the general public.
Each participant will receive modest financial compensation (100 CAD) for participation. For participants who drop out, payments will be pro-rated for the length of time they stayed in the study.
Assignment of interventions
Allocation and blinding
Participants will be randomly assigned to either the control or experimental group with a 1:1 allocation as per a computer generated randomisation schedule stratified by site and patient sex. To ensure allocation concealment, the randomisation will be handled by a member of staff not directly associated with recruitment, assessment or intervention delivery. The randomisation code will not be released until the patient has completed all baseline assessments. The staff member will provide a letter detailing the randomisation to the participant and will inform the interventionists of the patient's group allocation. None of the staff responsible for conducting assessments will be provided details of the allocation. In addition, all participants are asked to not indicate their assignment to any member of staff other than the interventionist they are working with. If a member of the assessment team is informed by the patient, they will remove themselves from any further assessment of that participant. As such, the participant and intervention staff will not be blinded to the group allocation, but the assessment staff will be.
Retention
Once randomised, a participant will be considered as being included in the study. Once all postintervention assessments have been completed (irrespective of adherence to the protocol), a participant will be consider to have completed the study. Based on our previous work with similar exercise interventions,29 we estimate that the rate of loss to follow-up will be 5%.
Participant withdrawal
Participants may withdraw from the study for any reason at any time. The senior investigator of the research project or the local ethics committee may withdraw the participant if new information or discoveries indicate that their participation in the project is not in their interest, if they do not follow the guidelines of the research project, or if there are administrative reasons to abandon the project.
Data management
Data forms and data entry
All data will be entered electronically using Microsoft Access. This will be carried out at either HSCM or MCI, depending on where the data originated. Original study forms will be entered and kept on file at the participating site. All data will be entered into two parallel electronic databases (double entry). Participant files are to be stored in numerical order and stored in a secure and accessible place and manner. Participant files will be maintained in storage for a period of 25 years after completion of the study. All blood and sputum samples will be stored in the HSCM tissue bank for an initial period of 5 years following analyses.
Data transmission and editing
The data entry screens will resemble the paper forms. Data integrity will be managed using referential data rules, valid values and using pull-down menus for certain categorical codes. Checks will be applied at the time of data entry into a specific field.
Data discrepancy inquiries
Additional errors will be detected by using SAS proc compared with detecting missing data or specific errors between the two electronically created datasets. These errors will be summarised along with detailed descriptions for each specific problem in Data Query Reports. The research assistant who receives the inquiry will respond by checking the original forms for inconsistency, checking other sources to determine the correction, modifying the original (paper) form and updating the two electronic datasets.
Security and back-up of data
All forms related to study data will be kept in locked cabinets. Access to the study data will be restricted. A complete back-up of the two electronic databases will be performed daily by the respective research centres. In addition, back-ups onto local external hard drives will be conducted weekly.
Study status reports
A monthly report on recruitment, study participations and completion will be provided to the study PI.
Proposed statistical analyses
Primary outcome
A general linear model will be used with the postintervention ACQ score as the dependent variable, treatment group as the between participant factor and age, baseline inhaled corticosteroid prescription (as a proxy of asthma severity), body weight change and pretreatment ACQ scores as covariates. Prior to conducting the analyses, preliminary examination of the assumptions of the analysis of covariance model will be conducted. Particular attention will be given to homogeneity of regression assumption, violations of heteroscedasticity of errors and non-linearity. Should these assumptions be violated, we will use interaction terms or appropriate transformation as necessary.
Secondary analyses
In parallel to the primary outcome, a series of general linear models will be used to assess intervention effects on ACT, AQLQ and the inflammatory markers. With the exception of substituting the pretreatment ACQ levels with the pretreatment levels of the dependent variable, all other covariates will be the same.
As defined by Baron et al70 ,71 and adapted by Kraemer et al72 ,73 for two group studies, a series of mediator analyses, with Sobel testing, will be conducted to explore inflammatory changes as a mechanism linking exercise to improved asthma control. To accomplish this, we will use a two-step regression model approach. In step 1, a simple general linear model (GLM) regression will be conducted with change in exercise (defined as the difference in postintervention VO2 minus preintervention VO2) as the independent variable and change in asthma control (defined as post minus preintervention levels) as the dependent variable. The above defined covariates, plus intervention group will be included. In step 2, the change in inflammatory markers, as well as the interaction between the change in VO2 and the change in inflammatory markers, will be included as additional independent variables, with the change in asthma control as the dependent variable and the above covariates. It should be noted that each inflammatory marker will be included in a separate set of analyses. In these kinds of analyses, a main or interaction effect of the inflammatory marker in step 2 would indicate that that particular marker mediated the relationship between exercise and asthma control.72 These kinds of analyses may not provide us with definitive causal and/or mechanistic relationships but will allow us to further expand our knowledge in the area.
p Value reporting will be consistent with the requirements of the place where the results will be published. Up-to-date versions of SAS (Cary, North Carolina, USA) will be used to conduct analyses. For all tests, we will use two-sided p values with an α≤0.05 level of significance. Multiple tests will be corrected with Benjamini and Hochberg's False Discovery Rate correction procedure.74–76
Analysis population and missing data
All main analyses will use intention-to-treat, considering all patients as randomised regardless of their adherence to the treatment protocol or completion of postintervention assessments. These analyses will be used to define the efficacy of aerobic exercise to influence asthma control. However, consistent with other behavioural trials, we will also conduct a series of completer analyses (ie, inclusion of only those with complete preintervention and postintervention assessment data). These will be conducted as quasisensitivity analyses. Given our expectation that very few patients will be lost to follow-up, these analyses should agree very closely.
Imputation procedure for missing data
We will report reasons for withdrawal for each randomisation group and compare the reasons qualitatively. Incomplete postintervention assessment data will be included in the analysis by modern imputation methods for missing data.77 ,78 The main feature of the approach is the creation of a set of clinically reasonable imputations for the respective outcome for each dropout. This will be accomplished using a set of repeated imputations created by predictive models based on the majority of participants with complete data. The imputation models will reflect uncertainty in the modelling process and inherent variability in patient outcomes, as reflected in the complete data. After the imputations are completed, all the data (complete and imputed) will be combined and the analysis performed for each imputed-and-completed dataset. Rubin's method of multiple (ie, repeated) imputation will be used to estimate the treatment effect. We propose to use five datasets. Analyses will follow Harrell's guidelines78 and will use the multiple imputation procedure available in SAS (SAS Inc, North Carolina, USA).
Data monitoring and interim analyses
Owing to the small size of the trial and the relatively short study duration, no data monitoring committee will be established and no interim analyses will be conducted.
Harm
In our study, an adverse event will be defined as any untoward medical occurrence in a participant without regard to the possibility of a causal relationship. Adverse events will be collected after the participant has provided consent and enrolled in the study. All adverse events occurring after entry (signing of a consent form) into the study and until completion of postintervention assessments or study withdrawal will be recorded. An adverse event that meets the criteria for a serious adverse event (SAE) will be reported to the local Research Ethics Board (REB) as an SAE. An SAE for this study is any untoward medical occurrence that is believed by the investigators to be causally related to participating in the study and results in any of the following: life-threatening condition (ie, immediate risk of death); severe or permanent disability or prolonged hospitalisation. SAEs occurring after a participant has discontinued from the study will NOT be reported unless the investigators feel that the event may have been caused by the study. Investigators will determine the relatedness of an event to the study intervention or procedure based on a temporal relationship, as well as whether the event is unexpected or unexplained given the participant's clinical course, previous medical conditions and concomitant medications. All SAEs will be reported for the trial and if there is sufficient power to assess intervention differences, a general linear model (see above) will be conducted.
Auditing
There will be no additional ongoing auditing of the data or trial conduct beyond that which has been detailed above.
Ethics and dissemination
Research ethics approval
The Ex-asthma study has been approved by the HSCM (ID: 2009–09–61) and McGill University Health Centre (ID: 10–076-BMC) REB. This approval covers the consent forms, all study procedures and assessments, recruitment scripts and advertising material. All study related documents, with the exception of the protocol, were approved in both French and English. The study complies with all REB policies and guidelines. Subsequent to the initial review and approval, the responsible local REB will review the protocol and progress reports (including safety) at least annually. In addition, within 12 months of study completion, the investigative team will provide each local REB with a final report.
Modifications of the protocol
Any modifications to the protocol which may impact on the conduct of the study, potential benefit of the patient or affect patient safety, including changes of study objectives, study design, patient population, sample sizes, study procedures or significant administrative aspects, will require a formal amendment to the protocol. Such amendment will be agreed upon by the investigative team and approved by the REBs prior to implementation.
Obtaining informed consent
Trained rsesearch assistants will introduce the trial to patients who will be shown a printed powerpoint presentation regarding the main aspects of the trial. They will discuss the trial with patients in light of the information provided. The patients will then be able to have an informed discussion with the participating consultant. Research assistants will obtain written consent from the patients willing to participate in the trial. All information and consent forms are provided in both French and English.
Confidentiality
All study-related information will be stored securely at the study sites. All participant information will be stored in locked file cabinets in areas with limited access. All laboratory specimens, reports, data collection, process and administrative forms will be identified by a coded identification number only to maintain participant confidentiality. All local databases will be secured with password-protected access systems.
Access to data
All final data will be stored centrally and all analyses will be conducted through the research team at HSCM. All investigators and students will have the capacity to request ancillary analyses.
Ancillary and post-trial care
No additional ancillary or post-trial care will be provided beyond the usual care covered by the Quebec provincial healthcare plan.
Knowledge translation strategy for trial results
The scientific integrity of the project requires that the data from all sites be analysed study-wide and reported as such. Substantive contributions to the design, conduct, interpretation and reporting of a clinical trial are recognised through the granting of authorship on the final trial report. The findings of this trial will be disseminated to a number of stakeholder groups.
The scientific and professional community, dissemination to this community will be performed through traditional channels of presentations at scientific meetings, grand rounds and publication of journal articles in open-access internet format as well as traditional publications.
Research participants, individuals who participate in the research will all receive personalised letters summarising their own personal results in the trial, as well as the overall results. These will be presented to participants at an end-of-study reception where printed results of the study will be distributed and explained.
General public, media will be invited to the participants’ reception to film and interview the researchers about the study results, as well as any participants who provide such consent. A study website has been created (accessible via the Montreal Behavioural Medicine Centre website (http://mbmc-cmcm.ca/en/projects/Ex-Asthma.php)) that describes the study and recruitment. On completion of the study, photographs of the end-of-study reception and press releases as well as downloads of scientific papers will be posted. The lay press will also be targeted to publish lay summaries of the study findings (table 1).
SPIRIT trial registration dataset80
Authorship policy
In general, named authorship for all secondary abstracts and papers will be defined as per the guidelines of the International Committee of Medical Journal Editors.79 Wherever possible, after the inclusion of all named authors, the author list will be completed with … “for the Ex-Asthma Study Group, with the full list of investigators provided in an appendix”.
Public access to the full protocol, participant-level dataset and statistical code
There are currently no plans to post the protocol, de-identified dataset or statistical code on a public access data archive for sharing purposes. However, the investigators would be happy to discuss data sharing with interested researchers.
References
Footnotes
-
Contributors All authors provided important contributions to the development of the protocol and revision of the current manuscript. Specifically, SLB is the study PI and lead writer. KLL was responsible for developing the protocol adherence strategy. JB is site PI for the Montreal Chest Institute. PE is site PI for the Jewish General Hospital. KM will conduct the immune function analyses. DG was responsible for helping develop aspects of study design and analytical strategies. ML is the medical director of the HSCM site. VP helped develop the exercise protocol. BKP provided critical input on the aspects of exercise immunology. Finally, all study authors will be actively involved in the interpretation of the final data and main study manuscript.
-
Funding Funding for the collection of data is being provided by operating grant MOP 93807 from the Canadian Institutes of Health Research (CIHR) and pilot data collection was made possible by funding from Concordia University. New investigator salary support was provided by the Fonds de la recherche du Québec: Santé (FRQS) and CIHR (SLB and KLL). The CIM (Centre of Inflammation and Metabolism) is supported by a grant from the Danish National Research Foundation (02-512-55: BKP). The study funders will have no role in study design; collection, management, analysis and interpretation of the data; writing of the report or the decision to submit the report for publication. The authority for these activities will solely rest with the study investigators.
-
Competing interests None.
-
Ethics approval Hopital du Sacre-Coeur de Montreal, McGill University Health Centre.
-
Provenance and peer review Not commissioned; internally peer reviewed.