Article Text

Abstract
Introduction Among critically ill patients undergoing orotracheal intubation in the emergency department (ED) or intensive care unit (ICU), failure to visualise the vocal cords and intubate the trachea on the first attempt is associated with an increased risk of complications. Two types of laryngoscopes are commonly available: direct laryngoscopes and video laryngoscopes. For critically ill adults undergoing emergency tracheal intubation, it remains uncertain whether the use of a video laryngoscope increases the incidence of successful intubation on the first attempt compared with the use of a direct laryngoscope.
Methods and analysis The DirEct versus VIdeo LaryngosCopE (DEVICE) trial is a prospective, multicentre, non-blinded, randomised trial being conducted in 7 EDs and 10 ICUs in the USA. The trial plans to enrol up to 2000 critically ill adults undergoing orotracheal intubation with a laryngoscope. Eligible patients are randomised 1:1 to the use of a video laryngoscope or a direct laryngoscope for the first intubation attempt. The primary outcome is successful intubation on the first attempt. The secondary outcome is the incidence of severe complications between induction and 2 min after intubation, defined as the occurrence of one or more of the following: severe hypoxaemia (lowest oxygen saturation <80%); severe hypotension (systolic blood pressure <65 mm Hg or new or increased vasopressor administration); cardiac arrest or death. Enrolment began on 19 March 2022 and is expected to be completed in 2023.
Ethics and dissemination The trial protocol was approved with waiver of informed consent by the single institutional review board at Vanderbilt University Medical Center and the Human Research Protection Office of the Department of Defense. The results will be presented at scientific conferences and submitted for publication in a peer-reviewed journal.
Trial registration number ClinicalTrials.gov Registry (NCT05239195).
- adult intensive & critical care
- statistics & research methods
- accident & emergency medicine
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
STRENGTHS AND LIMITATIONS OF THIS STUDY
This protocol describes in detail the design and methods for a large, pragmatic trial of laryngoscope type for the emergency tracheal intubation of critically ill adults.
Conduct in the emergency departments and intensive care units of multiple centres among operators with diverse prior experience with tracheal intubation, as well as broad patient eligibility criteria, will increase the external validity of trial results.
Patients, clinicians and investigators are not blinded to the study group assignment after randomisation.
Introduction
Tracheal intubation is a common procedure in the emergency department (ED) and intensive care unit (ICU). Among critically ill patients undergoing tracheal intubation, failure to intubate the trachea on the first attempt is associated with increased risk of complications, including hypoxaemia, hypotension, aspiration and cardiac arrest.1 2
Emergency tracheal intubation is typically performed in three discrete steps. First, the patient is administered medications to facilitate optimal intubating conditions (rapid sequence induction). Second, a clinician inserts a laryngoscope into the patient’s mouth to visualise the vocal cords (laryngoscopy). Third, an endotracheal tube is inserted into the mouth, alongside the laryngoscope, and the tube is advanced past the vocal cords into the trachea (intubation).
The direct laryngoscope, the traditional instrument consisting of a battery-containing handle attached to a blade with a light source, has been used to visualise the vocal cords for tracheal intubation for over 100 years and remains the most commonly used device for the intubation of critically ill adults in the ED or ICU.2–5 The operator uses the direct laryngoscope to displace the tongue and elevate the epiglottis to facilitate intubation of the trachea under direct visualisation. Obtaining an adequate view of the larynx with a direct laryngoscope can be challenging, especially for inexperienced operators. Once a view of the larynx is obtained, passage of the endotracheal tube follows the operator’s direct line of sight through the mouth to the vocal cords.
Over the last two decades, video laryngoscopes have provided an alternative to direct laryngoscopes for visualising the vocal cords to facilitate tracheal intubation.6 7 A camera embedded near the tip of the video laryngoscope blade transmits an image of the vocal cords to a screen that the operator can view during the procedure.8 Because the camera is located near the tip of the laryngoscope blade, obtaining a view of the larynx may be easier with a video laryngoscope compared with a direct laryngoscope. However, because this view can be obtained without generating a direct line of sight through the mouth to the vocal cords, the process of passing an endotracheal tube may be more difficult when using a video laryngoscope. When considering both aspects of tracheal intubation, visualising the vocal cords and passing the endotracheal tube, it remains uncertain whether the use of a video laryngoscope increases the incidence of successful intubation on the first attempt.
Among elective tracheal intubations in the operating room, the use of video laryngoscope probably increases the incidence of successful intubation on the first attempt and decreases complications compared with the use of a direct laryngoscope, supported with moderate certainty in the existing anaesthesiology literature.9 Extrapolating the results of randomised clinical trials conducted in the operating room to non-operating room settings is problematic because of factors related to the patient, the operator and the environment.10 11 Because tracheal intubation of critically ill adults outside of the operating room is common, complications of intubation in the ED and ICU are common, and the use of a video laryngoscope during intubation in the ED and ICU has increased significantly over time,9 12 understanding the effects of use of a video laryngoscope versus direct laryngoscope on successful intubation on the first attempt in these settings is a priority.
Previous trials randomising patients to the use of a video laryngoscope or a direct laryngoscope during emergency tracheal intubation in prehospital,13–18 ED19–25 and ICU settings26–32 have been small and heterogeneous and have generally suggested that while a video laryngoscope improves the view of the larynx and reduces the incidence of oesophageal intubation, it may not affect the incidence of successful intubation on the first attempt. Findings were similar in the largest such trial to date, a 371-patient, multicentre, randomised clinical trial in French medical ICUs in which the use of video laryngoscope failed to improve successful intubation on the first attempt (68% vs 70%; p=0.60) and was associated with a greater incidence of severe periprocedural complications in post-hoc analyses.33
The sample size of these prior trials did not provide sufficient statistical power to definitively rule out a clinically important effect of the use of a video laryngoscope versus direct laryngoscope on successful intubation on the first laryngoscopy attempt or the incidence of complications. To compare the effectiveness of these two commonly used devices during this important emergency procedure, a large trial conducted across a wide variety of clinical settings, operator specialties and levels of operator experience is required. Therefore, we designed the DirEct versus VIdeo LaryngosCopE (DEVICE) trial to test the hypothesis that, among critically ill adults undergoing emergency tracheal intubation in the ED or ICU, the use of a video laryngoscope will increase the incidence of successful intubation on the first attempt compared with the use of a direct laryngoscope.
Methods and analysis
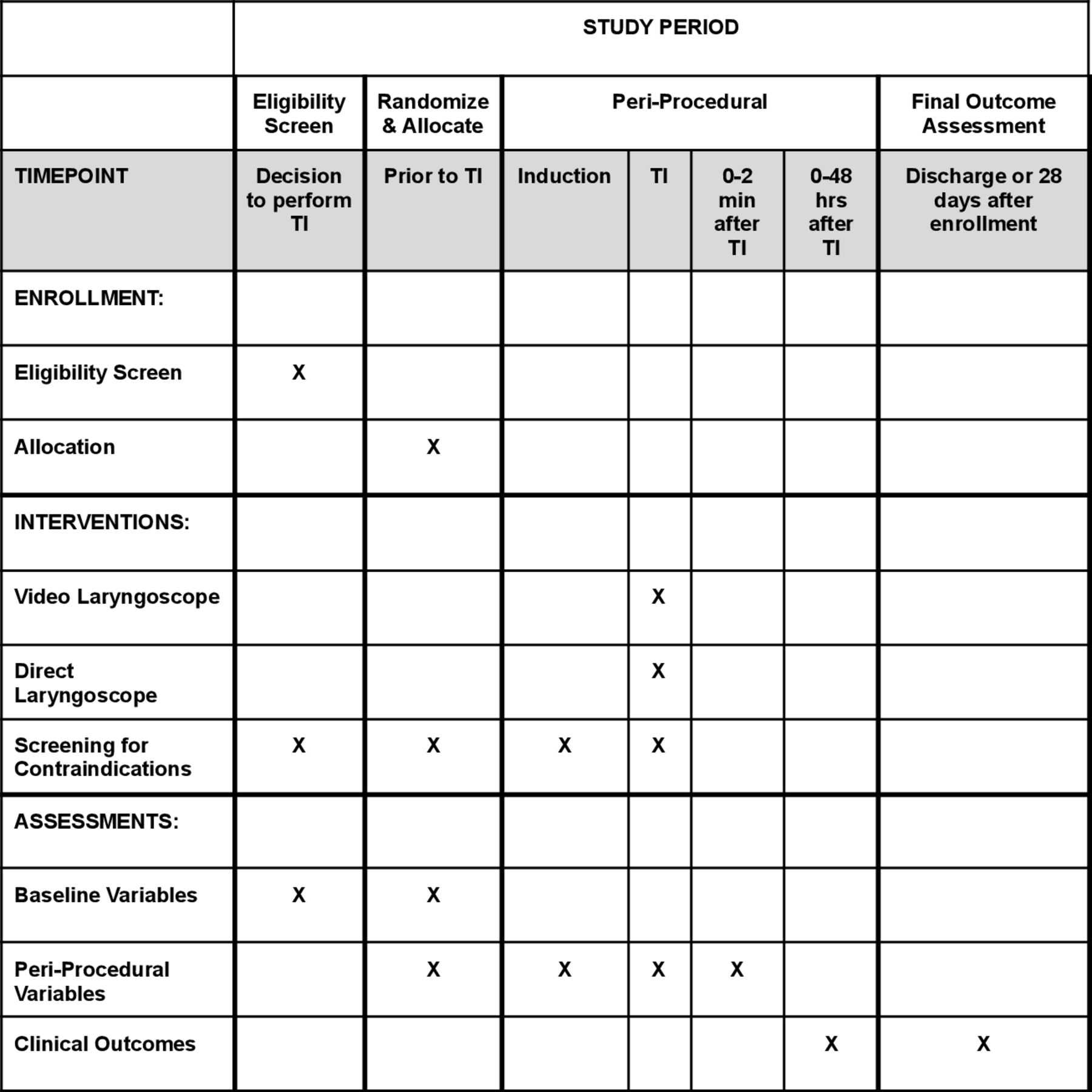
This manuscript was written in accordance with Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) guidelines (figure 1 and online supplemental file 1).34
Supplemental material
{kind=link}
Schedule of enrolment, interventions and assessments in the DEVICE trial. DEVICE, DirEct versus VIdeo LaryngosCopE; TI, tracheal intubation.
Patient and public involvement
Materials used to communicate details of the study with patients and family members were developed with input from the Vanderbilt Community Advisory Council. Study authors will disseminate the results of this study online and via social media in forms suitable for public understanding.
Study design
The DEVICE trial is a pragmatic, multicentre, unblinded, parallel-group, randomised trial comparing the use of a video laryngoscope with the use of a direct laryngoscope for the first attempt at emergency tracheal intubation among critically ill adults in the ED and ICU. The primary outcome is successful intubation on the first attempt. An independent data and safety monitoring board (DSMB) is monitoring the progress and safety of the trial. Study institutions and investigators are listed in the online supplemental file 2.
Study population
The inclusion criteria for this study are:
Patient is located in a participating unit.
Planned procedure is orotracheal intubation using a laryngoscope.
Planned operator is a clinician expected to routinely perform tracheal intubation in the participating unit.
The exclusion criteria for the study are:
Patient is known to be less than 18 years old.
Patient is known to be pregnant.
Patient is known to be a prisoner.
Immediate need for tracheal intubation precludes safe performance of study procedures.
Operator has determined that the use of a video laryngoscope or use of a direct laryngoscope is required or contraindicated for the optimal care of the patient.
Randomisation and treatment allocation
Patients are randomised in a 1:1 ratio to undergo intubation using a video laryngoscope or using a direct laryngoscope for the first attempt in permuted blocks of variable size, stratified by study site. Study group assignments are generated using a computerised randomisation sequence, placed in sequentially numbered opaque envelopes and distributed to enrolling sites. Before opening the envelope, the operator determines that the patient meets eligibility criteria, records the predicted difficulty of intubation (‘easy’, ‘moderate’ or ‘difficult’) and selects the blade shape the operator plans to use if the patient is randomised to the video laryngoscope group (‘hyperangulated’ or ‘non-hyperangulated/standard geometry’). The operator or delegate then opens the envelope. Patients are enrolled once the envelope is opened to reveal the study group assignment. After enrolment and randomisation, patients, treating clinicians and study personnel are not blinded to study group assignment.
Study interventions
Video laryngoscope group
For patients assigned to the video laryngoscope group, operators are instructed to use a video laryngoscope on the first laryngoscopy attempt. A video laryngoscope is defined as a laryngoscope with a camera and a video screen. Trial protocol does not dictate the brand of video laryngoscope or the geometry of the laryngoscope blade (eg, hyperangulated vs non-hyperangulated), but these details will be recorded. Operators are encouraged, but not required, to view the video screen during laryngoscopy (‘indirect laryngoscopy’) and tracheal intubation.
Direct laryngoscope group
For patients assigned to the direct laryngoscope group, operators are instructed to use a direct laryngoscope on the first laryngoscopy attempt. A direct laryngoscope is defined as a laryngoscope without a camera and a video screen. Trial protocol does not dictate the brand of direct laryngoscope or the geometry of the laryngoscope blade (eg, curved (Macintosh) vs straight (Miller)), but these details will be recorded.
Co-interventions and subsequent attempts at laryngoscopy and intubation
Study group assignment determines only the type of laryngoscope (video vs direct) used on the first laryngoscopy attempt. If determined to be required to ensure optimal care of the patient, treating clinicians may use any device at any time, regardless of study group assignment. Cases in which clinicians use a laryngoscope discordant with randomised assignment on the first intubation attempt will be documented and tracked. All aspects of the intubation procedure, except the type of laryngoscope used on the first attempt, are at the discretion of treating clinicians, including selection of sedative and neuromuscular blocking medications, patient positioning, approach to preoxygenation, use of a bougie or a stylet, and endotracheal tube size. Best practices in tracheal intubation will be encouraged according to clinical protocols at the study sites. The trial intervention ends after the first attempt at laryngoscopy. If the first attempt is unsuccessful, the operator may use any method of intubation on subsequent intubation attempts, including the use of a direct laryngoscope in the video laryngoscope group or use of a video laryngoscope in the direct laryngoscope group. The type of laryngoscope used during the initial and final laryngoscopy attempt will be collected and reported.
Data collection
A trained observer, not directly involved with the intubation procedure, collects data for key periprocedural outcomes. These outcomes include successful intubation on the first attempt, time interval between laryngoscopy and successful intubation, the oxygen saturation (SpO2) and systolic blood pressure at induction, the lowest SpO2 and systolic blood pressure between induction and 2 min after successful intubation, and new or increased vasopressor administration between induction and 2 min after successful intubation. Observers may be clinical personnel on the enrolling unit (eg, physician, nurse or pharmacist) or research study personnel.
Immediately following the intubation procedure, the operator completes a paper data collection form to record the approach to preoxygenation, oxygenation and ventilation between induction and laryngoscopy, the brand of laryngoscope used, the blade shape, the Cormack-Lehane grade of laryngeal view,35 use of the video screen to visualise the larynx (if applicable), use of a bougie or a stylet, reasons for failure to intubate on the first attempt (if applicable), intubation approaches on subsequent attempts, difficult airway characteristics observed before or during the procedure (facial trauma, small mouth opening, limited neck mobility, cervical collar, large neck, obesity, fluids obscuring view of vocal cords, upper airway obstruction or oedema), and complications of intubation (witnessed pulmonary aspiration, oesophageal intubation, injury to airways, injury to teeth, cardiac arrest between induction and 2 min following intubation). The diagnosis of oesophageal intubation is made by the operator based on the presence of any clinical sign including visual inspection, capnography, or absence of breath sounds or chest rise. Operators also record their specialty, training level, and estimates of the number of previous intubations they have performed and the number of previous intubations they have performed using a direct laryngoscope.
Study personnel at each site review the medical record to collect data on baseline patient characteristics, pre-laryngoscopy and post-laryngoscopy management, and clinical outcomes at 28 days after enrolment.
The following variables are collected:
Baseline: age, sex, height, weight, race, ethnicity, Acute Physiology and Chronic Health Evaluation II score,36 active medical problems at the time of enrolment, comorbidities, indication for intubation, vasopressor receipt in the hour prior to enrolment, highest fractional inspired oxygen (FIO2) in the hour prior to enrolment, lowest SpO2/FIO2 (or arterial oxygen pressure/FIO2) ratio in the hour prior to enrolment, pre-procedural Glasgow Coma Scale score,37 oxygen delivery device at enrolment, assessment of the likelihood of a difficult intubation, presence of difficult airway characteristics (limited mouth opening, small mandible, large tongue, short neck, large neck circumference, limited anatomical neck mobility, cervical immobilisation due to trauma, obesity), operator’s level of training and specialty, operator’s prior intubation experience.
Periprocedural: lowest SpO2 from enrolment to induction, approach to and duration of preoxygenation, time of sedative administration, sedative agent and dose administered, neuromuscular blocking agent and dose administered, SpO2 and systolic blood pressure at the time of induction, approach to oxygen administration and ventilation between induction and the first attempt at laryngoscopy, time of start of first laryngoscopy attempt, laryngoscope used on first attempt (model, blade size, blade shape), use of video screen (if applicable) on the first laryngoscopy attempt, best Cormack-Lehane grade of view35 on the first laryngoscopy attempt, presence of body fluid obstructing view of the larynx, presence of upper airway obstruction or oedema, number of intubation attempts (number of times the laryngoscope entered the mouth, number of times the bougie entered mouth (if applicable), number of times the endotracheal tube entered the mouth), reason for failure of the first intubation attempt (if applicable), procedural adjustments made for the final intubation attempt, oesophageal intubation, injury to teeth, operator-reported pulmonary aspiration between induction and intubation, time of successful tracheal intubation, endotracheal tube size, lowest SpO2 from induction until 2 min after intubation, lowest systolic blood pressure from induction until 2 min after intubation, new or increased vasopressor administration from induction until 2 min after intubation, cardiac arrest from induction until 2 min after intubation not resulting in death within 1 hour of induction, cardiac arrest from induction until 2 min after intubation resulting in death within 1 hour of induction.
Twenty-four hours after enrolment: new pneumothorax detected in the first 24 hours after induction, vasopressor receipt at 24 hours after induction, SpO2 at 24 hours after induction, FIO2 at 24 hours after induction, positive end-expiratory pressure at 24 hours after induction, systolic blood pressure at 24 hours after induction.
In-hospital outcomes: ventilator-free days in the first 28 days, ICU-free days in the first 28 days and in-hospital mortality at 28 days. Definitions for ICU-free days and ventilator-free days are provided in the online supplemental files 3 and 4.
Primary outcome
The primary outcome is successful intubation on the first attempt. Successful intubation on the first attempt is defined as placement of an endotracheal tube in the trachea following a single insertion of a laryngoscope blade into the mouth and either a single insertion of an endotracheal tube into the mouth or a single insertion of a bougie into the mouth followed by a single insertion of an endotracheal tube into the mouth.
Data for the assessment of the primary outcome are collected by a trained independent observer using a structured data collection form that records the number of insertions of the laryngoscope blade, bougie (if used) and endotracheal tube into the patient’s mouth. In the event that data from the independent observer are missing, data from the operator’s self-report of successful intubation on the first attempt will be used.
Secondary outcome
The secondary outcome is the incidence of severe complications occurring between induction and 2 min following successful intubation. Severe complications are defined as one or more of the following:
Severe hypoxaemia (lowest SpO2 measured by pulse oximetry <80%).
Severe hypotension (systolic blood pressure <65 mm Hg or new or increased vasopressor administration).
Cardiac arrest not resulting in death.
Cardiac arrest resulting in death.
Cardiac arrest will be considered to have resulted in death if a patient who experienced cardiac arrest between induction and 2 min after intubation died within the 1 hour following intubation.
Exploratory outcomes
Exploratory procedural outcomes are as follows:
Duration of laryngoscopy and tracheal intubation. This is defined as the interval (in seconds) between the first insertion of a laryngoscope blade into the mouth and the final placement of an endotracheal tube or tracheostomy tube in the trachea.
Number of laryngoscopy attempts.
Number of attempts to cannulate the trachea with a bougie or endotracheal tube.
Successful intubation on the first attempt without a severe complication.
Reasons for failure to intubate the trachea on the first attempt, which include:
Inadequate view of the larynx.
Inability to intubate the trachea with an endotracheal tube.
Inability to cannulate the trachea with a bougie.
Attempt aborted due to a change in patient condition (eg, worsened hypoxaemia, hypotension, bradycardia, vomiting, bleeding).
Technical failure of the laryngoscope (eg, battery, light source, camera, screen).
Other.
Exploratory safety outcomes are as follows:
Oesophageal intubation.
Injury to the teeth.
Operator-reported aspiration.
Exploratory clinical outcomes are as follows:
ICU-free days in the first 28 days.
Ventilator-free days in the first 28 days.
28-day all-cause in-hospital mortality.
Sample size estimation
The minimum clinically important difference in successful intubation on the first attempt that would be needed to justify routine use of a video laryngoscope rather than a direct laryngoscope in the ED and ICU is uncertain. The current trial is designed to detect a 5% absolute difference between groups in the incidence of successful intubation on the first attempt. An absolute difference of 5% in successful intubation on the first attempt is similar to or smaller than the difference used in the design of prior airway management trials and is considered by airway management experts to be clinically meaningful.21 28 38 39 Assuming (1) an incidence of successful intubation on the first attempt of 80% in the direct laryngoscope group, (2) 90% statistical power, (3) a two-sided alpha of 0.05 and (4) enrolment at 16 sites with an intracluster correlation for the primary outcome of 0.05, we calculated that detecting a 5% absolute increase in the incidence of successful intubation on the first attempt would require enrolment of 1920 patients (960 per group). Anticipating missing data for up to 4% of enrolled patients, we will plan to enrol a total of 2000 patients (1000 per group).
DSMB and interim analysis
A DSMB composed of experts with backgrounds in emergency medicine, pulmonary and critical care medicine, anaesthesiology, bioethics and biostatistics has overseen the design of the trial and is monitoring its conduct. The DSMB will review a single interim analysis prepared by the study biostatistician at the anticipated halfway point of the trial, after enrolment of 1000 patients. The stopping boundary for efficacy was prespecified as a p value of 0.001 or less, using a Χ2 test, for the difference in the incidence of the primary outcome between groups. This conservative Haybittle-Peto boundary was selected to allow the final analysis to be performed using an unchanged level of significance (p<0.05). The DSMB retains the authority to stop the trial at any point, request additional data or interim analyses, or request modifications of the study protocol to protect patient safety. Trial safety monitoring and handling of adverse events are described in detail in the online supplemental file 5. Patient privacy and data storage details are listed in the online supplemental file 6.
Statistical analysis principles
Analyses will be conducted following reproducible research principles using R (R Foundation for Statistical Computing, Vienna, Austria).40 We will present summary tabulations by treatment group. For categorical variables, the number and proportion of patients will be presented. For continuous variables, the mean and SD or median and IQR will be presented, as appropriate.
We will analyse a single prespecified primary outcome and a single prespecified secondary outcome using a Χ2 test. Consistent with recommendations of the Food and Drug Administration41 and the European Medicines Agency,42 each will be tested using a two-sided p value with a significance level of 0.05 with contextual information provided via effect size and 95% CIs. The primary analysis will occur in an intent-to-treat fashion among all patients randomised, excluding only those patients whose data were withdrawn from the study. For all other analyses except safety analyses, emphasis will be placed on the estimate of effect size with 95% CIs, as recommended by the International Committee of Medical Journal Editors,43 and no corrections for multiple comparisons will be performed.
Main analysis of the primary outcome
The main analysis will be an unadjusted, intention-to-treat comparison of successful intubation on the first attempt between patients randomised to the video laryngoscope group and patients randomised to the direct laryngoscope group, using a Χ2 test. The difference in proportions, the associated 95% CI and a p value for the primary outcome will be presented.
Secondary analyses of the primary outcome
Multivariable modelling to account for covariates
To account for relevant covariates, we will develop a generalised linear mixed-effects model using a logit link function with the primary outcome as the dependent variable, study site as a random effect, and fixed effects of study group and the following prespecified baseline covariates: age, sex, body mass index, operator experience quantified as the operator’s total number of prior intubations and location of intubation (ED vs ICU). All continuous variables will be modelled assuming a non-linear relationship to the outcome using restricted cubic splines with between 3 and 5 knots.
Effect modification
We will examine whether prespecified baseline variables modify the effect of study group assignment (video laryngoscope vs direct laryngoscope) on the primary outcome using a formal test of statistical interaction in a generalised linear mixed-effects model with the primary outcome as the dependent variable, study site as a random effect and fixed effects of study group, the prespecified proposed effect modifier and the interaction between the two. For categorical variables, we will present the OR and 95% CIs within each prespecified subgroup. Continuous variables will not be dichotomised for analysis of effect modification but may be dichotomised for data presentation. In accordance with the Instrument for assessing the Credibility of Effect Modification Analyses recommendations,44 we have prespecified the following limited number of baseline variables as potential effect modifiers and the hypothesised direction of effect modification for each:
Patient location (ED vs ICU). We hypothesise that patient location will not modify the effect of study group assignment on the primary outcome.
Traumatic injury (yes vs no). We hypothesise that traumatic injury will modify the effect of study group assignment on the primary outcome, with a greater increase in the incidence of successful intubation on the first attempt with the use of a video laryngoscope compared with a direct laryngoscope among patients with traumatic injury compared with patients without traumatic injury.
Body mass index (kg/m2). We hypothesise that body mass index will modify the effect of study group assignment on the primary outcome, with a greater increase in the incidence of successful intubation on the first attempt with the use of a video laryngoscope compared with a direct laryngoscope among patients with higher body mass index as compared with patients with lower body mass index. This hypothesis of effect modification is supported by a non-significant trend toward effect modification in a meta-analysis of multiple prior randomised trials.9
Operator’s pre-enrolment assessment of the anticipated difficulty of intubation (easy; moderate; difficult; not recorded). We hypothesise that the operator’s pre-enrolment assessment will modify the effect of study group assignment on the primary outcome, with a greater increase in the incidence of successful intubation on the first attempt with the use of a video laryngoscope compared with a direct laryngoscope among patients assessed as difficult or moderate compared with easy. This hypothesis of effect modification is supported by significant effect modification in a meta-analysis of multiple prior randomised trials.9
Operator experience at the time of enrolment.
Total number of previous intubations performed by operator. We hypothesise that the total number of previous intubations performed by the operator will modify the effect of study group assignment on the primary outcome, with a greater increase in the incidence of successful intubation on the first attempt with the use of a video laryngoscope compared with a direct laryngoscope among operators with fewer previous intubations compared with operators with a greater number of previous intubations. This hypothesis of effect modification is supported by significant effect modification observed in a prior randomised trial among critically ill adults, but differs from a meta-analysis including trials of intubation in the operating room that did not observe effect modification based on the operator’s prior experience.9 28
Proportion of previous intubations performed by the operator using a direct laryngoscope. We hypothesise that the proportion of previous intubations performed by the operator using a direct laryngoscope will modify the effect of study group assignment on the primary outcome, with a greater increase in the incidence of successful intubation on the first attempt with the use of a video laryngoscope compared with a direct laryngoscope among operators with a lower proportion of previous intubations performed by the operator using a direct laryngoscope compared with operators with a higher proportion of previous intubations performed by the operator using a direct laryngoscope.
We will also perform an effect modification analysis for the primary outcome that includes a three-way interaction between study group, total number of previous intubations performed by the operator and proportion of previous intubations performed by the operator using a direct laryngoscope.
Sensitivity analyses of the primary outcome
We will assess the robustness of the findings of the primary analysis in a number of sensitivity analyses. First, because operators may choose to deviate from the assigned laryngoscope for the safety of the patient, we will repeat the primary analysis, but will consider patients for whom the operator crossed over on the first attempt from the assigned laryngoscope type to the non-assigned laryngoscope type not to have experienced successful intubation on the first attempt. Second, we will repeat the primary analysis among only patients for whom data on the primary outcome from the independent observer are available (ie, excluding cases in which operator self-report was the sole source of information for the primary outcome). Third, because the operator’s prior experience with each type of laryngoscope may affect the likelihood of success with a video laryngoscope compared with a direct laryngoscope, we will repeat the primary analysis among only cases in which the proportion of prior intubations the operator has performed using a direct laryngoscope is between 0.25 and 0.75.
Analysis of the secondary outcome
For the secondary outcome, severe complications occurring between induction and 2 min following intubation, we will perform an unadjusted, intention-to-treat comparison of patients randomised to the video laryngoscope group versus patients randomised to the direct laryngoscope group, using a Χ2 test.
Analyses of exploratory outcomes
For all prespecified exploratory outcomes, we will conduct unadjusted, intention-to-treat analyses comparing patients randomised to the video laryngoscope group versus patients randomised to the direct laryngoscope group. We will calculate absolute risk differences or differences in medians between groups with the associated 95% CIs.
Handling of missing data
We anticipate that no data on the primary outcome will be missing. When data are missing for the secondary or exploratory outcomes, we will perform complete case analysis, excluding cases where the data for the analysed outcome are missing. There will be no imputation of missing data for these outcomes. In adjusted analyses, missing data for covariates will be imputed using multiple imputations.
Trial status
The DEVICE trial is a prospective, multicentre, non-blinded randomised clinical trial comparing the use of a video laryngoscope with the use of a direct laryngoscope for the first attempt at tracheal intubation of critically ill adults in the ED and ICU. Patient enrolment began on 19 March 2022 and is being conducted in 7 EDs and 10 ICUs in the USA.
Ethics and dissemination
Waiver of informed consent
Critically ill patients undergoing tracheal intubation in the ED or ICU are at significant risk of morbidity and mortality from their underlying illness. Most patients undergoing tracheal intubation in routine clinical care are intubated using either a video laryngoscope or a direct laryngoscope on the first attempt. Any benefits or risks of these two approaches are experienced by patients undergoing tracheal intubation in clinical care, outside the context of research. As a requirement for enrolment in the DEVICE trial, the patient’s treating clinician must believe that either a video laryngoscope or a direct laryngoscope would be a safe and reasonable approach for the patient (otherwise the patient is excluded). Therefore, making the decision between the two approaches randomly (by study group assignment) rather than by a clinician who thinks either approach is safe and reasonable for the patient is expected to pose no more than minimal additional risk.
Obtaining informed consent for participation in the study would be impracticable. The majority of patients undergoing emergency tracheal intubation lack decisional capacity due to their underlying critical illness and surrogate decision-makers are frequently absent. Further, emergency tracheal intubation is a time-sensitive procedure with only minutes between the decision to perform intubation and the completion of the procedure. Meaningful informed consent could not be executed in this brief window and attempting to obtain informed consent would lead to potentially deleterious and unethical delays in intubation which would increase the risk of hypoxaemia, hypotension and periprocedural cardiac arrest.
Because the study involves minimal incremental risk, the study would not adversely affect the welfare or privacy rights of the participant and obtaining informed consent would be impracticable, a waiver of informed consent was requested from and approved by the single institutional review board (IRB) at Vanderbilt University Medical Center (reference number 211272). This is consistent with previous randomised trials comparing alternative approaches with tracheal intubation commonly used in clinical care.28 38 39 45–50 This approach was approved by the US Department of Defense Defense Health Agency Human Research Protection Office (EIRB# 944893). IRBs at participating sites reviewed the protocol, addressed any local contextual factors with the site principal investigator, and ceded responsibility for ethics approval and study oversight to the single IRB.
Information for patients and families
Information regarding the study is made available to patients and families using a patient and family information sheet. The patient and family information sheet contains information on the purpose of the trial, study procedures, risks and discomforts, benefits, use of protected health information, confidentiality and investigator contact information. The Defense Health Agency Human Research Protection Office determined that this procedure meets the requirements of 32 CFR 219 and DODI 3216.02_AFI40-402. At centres with a significant population of non-English-speaking patients, the patient and family information sheet has been translated into Spanish and Somali languages and is made available to those patients.
Protocol changes
Any further amendments to the protocol will be recorded on ClinicalTrials.gov as per SPIRIT guidelines. See the online supplemental file 7 for details on how protocol changes will be handled.
Dissemination plan
Trial results will be submitted to a peer-reviewed journal and will be presented at one or more scientific conferences.
Ethics statements
Patient consent for publication
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @drstrent, @SGandotraMD, @alatimer13, @BradLloydVFL
JDC and MWS contributed equally.
Contributors Study concept and design—MEP, BED, SAT, SGS, BJL, DJ, WHS, TWR, AAG, JDC and MWS. Acquisition of data—MEP, BED, SAT, DR-A, KS, DWR, SG, JPG, KWG, AL, MRW, SG, DJV, JPW, SJH, ISD, CRB, VK, JJB, BDL, SWR, JAP, SM, DBP, HDW, AE, CH, AMJ, JTH, AAG, JDC and MWS. Drafting of the manuscript and study supervision—MEP, BED, SAT, JDC and MWS. Approval of the final version of this manuscript and critical revision of the manuscript for important intellectual content—MEP, BED, SAT, DR-A, KS, DWR, SG, JPG, KWG, AL, MRW, SG, DJV, JPW, SJH, ISD, CRB, VK, JJB, BDL, SWR, JAP, SM, DBP, HDW, AE, CH, AMJ, JTH, SGS, BJL, BI, LW, JPR, KNW, DJ, WHS, TWR, AAG, JDC and MWS.
Funding The research was funded in part by the US Department of Defense, Defense Health Agency, J9 Office, RESTORAL programme. Data collection used the Research Electronic Data Capture (REDCap) tool developed and maintained with Vanderbilt Institute for Clinical and Translational Research grant support (UL1 TR000445 from NCATS/NIH).
Disclaimer The funding institutions had no role in (1) conception, design or conduct of the study; (2) collection, management, analysis, interpretation or presentation of the data; or (3) preparation, review or approval of the manuscript. The views expressed are those of the authors and do not reflect the official views or policy of the Department of Defense or its components.
Competing interests MWS was supported by the National Heart, Lung and Blood Institute (K23HL143053). JDC was supported by the National Heart, Lung and Blood Institute (K23HL153584-01). JPG received support from the CHEST Foundation for instruction and travel. TWR was supported in part by the National Institutes of Health and received consulting payments from Cumberland Pharmaceuticals and Cytovale, and served on a data safety and monitoring board for Sanofi.
Patient and public involvement Patients and/or the public were involved in the design, or conduct, or reporting, or dissemination plans of this research. Refer to the Methods section for further details.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.