Article Text

Abstract
Introduction Supplemental oxygen is commonly used in trauma patients, although it may lead to hyperoxaemia that has been associated with pulmonary complications and increased mortality. The primary objective of this trial, TRAUMOX2, is to compare a restrictive versus liberal oxygen strategy the first 8 hours following trauma.
Methods and analysis TRAUMOX2 is an investigator-initiated, international, parallel-grouped, superiority, outcome assessor-blinded and analyst-blinded, randomised, controlled, clinical trial.
Adult patients with suspected major trauma are randomised to eight hours of a restrictive or liberal oxygen strategy. The restrictive group receives the lowest dosage of oxygen (>21%) that ensures an SpO2 of 94%. The liberal group receives 12–15 L O2/min or FiO2=0.6–1.0.
The primary outcome is a composite of 30-day mortality and/or development of major respiratory complications (pneumonia and/or acute respiratory distress syndrome).
With 710 participants in each arm, we will be able to detect a 33% risk reduction with a restrictive oxygen strategy if the incidence of our primary outcome is 15% in the liberal group.
Ethics and dissemination TRAUMOX2 is carried out in accordance with the Helsinki II Declaration. It has been approved by the Danish Committee on Health Research Ethics for the Capital Region (H-21018062) and The Danish Medicines Agency, as well as the Dutch Medical Research Ethics Committee Erasmus MS (NL79921.078.21 and MEC-2021-0932). A website (www.traumox2.org) is available for updates and study results will be published in an international peer-reviewed scientific journal.
Trial registration numbers EudraCT 2021-000556-19; NCT05146700.
- Adult anaesthesia
- Adult intensive & critical care
- TRAUMA MANAGEMENT
- Respiratory physiology
Data availability statement
No data are available. NA.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
STRENGTHS AND LIMITATIONS OF THIS STUDY
TRAUMOX2 is an investigator-initiated, international, multicentre, parallel-grouped, superiority, outcome assessor-blinded and analyst-blinded, randomised, controlled, clinical trial.
The oxygen treatment in current trauma management is challenged in this trial with trauma patients being randomised to two different oxygen strategies in the initial period post-trauma.
The intervention is open label, and the assessment of the primary outcome is blinded.
The international setup will allow clinically relevant and generalisable results.
Oxygen delivered to the tissues is dependent on other factors not directly accounted for in this trial, such as cardiac output and blood loss.
Introduction
Background and rationale
In trauma resuscitation, supplemental oxygen is often administered to treat or prevent hypoxaemia as recommended by the Advanced Trauma Life Support manual.1 This guideline does not have a specific therapeutic goal. Oxygen is administered in many other situations too, sometimes in a non-consistent manner2–4 and very often without even being prescribed.5 In a recent systematic review, our group found the evidence for the use of supplemental oxygen in the trauma population to be sparse.6 Nevertheless, in a large retrospective study on 864 340 trauma patients, a propensity score matching analysis showed that the administration of supplemental oxygen was associated with a significantly increased risk of in-hospital mortality and acute respiratory distress syndrome (ARDS).7 Furthermore, a recent systematic review and meta-analysis comparing liberal versus restrictive oxygen strategy for a broad mix of acutely ill medical and surgical patients found an association between liberal oxygen administration and increased mortality.8 Of note, only one small study on trauma patients (patients with traumatic brain injury), which did not report mortality data, was included. Conversely, this study showed that degree of disability was significantly reduced at six months in the group receiving liberal compared with restrictive oxygen.9
Hyperoxaemia is a common finding in trauma patients5 10 and in mechanically ventilated patients in general.11 12 However, in the Intensive Care Unit (ICU) and in surgical patients, hyperoxaemia has been associated with major pulmonary complications.13 14 For example, a recent retrospective study found hyperoxaemia to be an independent risk factor for ventilator associated pneumonia.14 Nevertheless, a highly debated recommendation from the WHO states that adult patients undergoing general anaesthesia for surgical procedures should receive an FiO2 of 0.80 intraoperatively as well as in the immediate postoperative period for 2–6 hours to reduce the risk of surgical site infection.15 A randomised study, however, found no difference in the risk of surgical site infection according to FiO2 concentration intraoperatively.16 Furthermore, a study on 152 000 mechanically ventilated patients found no association between hyperoxia and mortality during the first 24 hours in the ICU,17 and another study on 14 000 mixed ICU patients found that a PaO2 of approximately 18 kPa resulted in the lowest mortality.18 Finally, a recent study randomised 2928 ICU patients to either low or high arterial oxygen tension target (defined as 8 vs 12 kPa), for a maximum of 90 days and found no difference in mortality.19
Therefore, whether the trauma population could benefit from a more restrictive supplemental oxygen approach than recommended by current international trauma guidelines presents a large and important knowledge gap. In a recent pilot randomised clinical trial (TRAUMOX1,20 Clinicaltrials.gov Registration number: NCT03491644), we compared a restrictive and a liberal oxygen strategy for 24 hours after trauma (n=41) and found maintenance of normoxaemia following trauma using a restrictive oxygen strategy to be feasible. The study served as the basis for the current larger clinical trial; TRAUMOX2.
In TRAUMOX2, we hypothesise that a restrictive compared with a liberal oxygen strategy for eight hours following trauma will result in a lower rate of 30-day mortality and/or major respiratory complications (pneumonia and/or ARDS) (combined endpoint).
Methods
Study setting
The protocol has been written according to the Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) 2013 statement with version 1.6.21 The SPIRIT 2013 checklist is presented in online supplemental appendix 1.
Supplemental material
TRAUMOX2 is an investigator-initiated, international, multicentre, parallel-grouped, superiority, outcome assessor-blinded and analyst-blinded, randomised, controlled, clinical trial.
Participating trauma centres must be able to provide definitive treatment of trauma patients (tertiary care, ie, no transfer to more specialised institution needed), possess a trauma registry and have an average of approximately 400 trauma patients per year. All trauma centres must be within the EU and thus the EU Clinical Trial Regulation are applied. A complete list of the study sites can be viewed on the trial website (www.traumox2.org).
Eligibility criteria
Patients aged 18 years or older, including fertile women, are included. Participants must have undergone blunt or penetrating trauma, be directly transferred from the scene of accident to one of the participating trauma centres and trigger trauma team activation (no secondary transfers from other hospitals). Moreover, the including physician must initially expect a hospital length of stay (HOS LOS) for 24 hours or longer. Patients with no/minor injuries after secondary survey are excluded if they are expected to be discharged within 24 hours to ensure only patients with a substantial trauma are included. Patients in cardiac arrest before or on admission and patients with a suspicion of carbon monoxide intoxication are excluded.
Interventions
Participants are randomised to eight hours of either restrictive or liberal supplemental oxygen treatment. The restrictive group receives the lowest dosage of oxygen (≥21%) that ensures an arterial oxyhaemoglobin saturation (SpO2, measured by pulse oximetry) target of 94% either using no supplemental oxygen, a nasal cannula, a non-rebreather mask or mechanical ventilation (intubated patients). Thus, only trial participants without a need for supplemental oxygen to maintain an SpO2≥94% can saturate above 94%. The liberal group receives 15 L O2/min via a non-rebreather mask for non-intubated trial participants and an FiO2=1.0 for intubated trial patients in the prehospital setting, in the trauma bay, and during intrahospital transportation. In the operating room, ICU, postanaesthesia care unit and ward the flow/FiO2 may be reduced to 12 L O2/min/FiO2=0.6 if the arterial oxygen saturation ≥98%. In both groups, preoxygenation should be done prior to intubation.
Outcomes
The primary outcome is a composite of 30-day mortality and/or development of major respiratory complications (pneumonia and/or ARDS). Secondary outcomes include mortality at 30 days and 12 months post-trauma, major respiratory complications (pneumonia and ARDS) within 30 days, HOS LOS, ICU length of stay (ICU LOS), days alive outside the ICU, time on mechanical ventilation (until 30 days), days alive without mechanical ventilation, reintubation within 30 days, pneumonia post-discharge, surgical site infections within 30 days, episodes with hypoxaemia during intervention (SpO2<90%) and EQ-5D-5L score at six and 12 months post-trauma and Glasgow Outcome Scale-Extended (GOSE) at six and 12 months post-trauma. The EQ-5D-5L score is a widely used generic measure for health-related quality of life and22 GOSE assesses physical and mental consequences of traumatic brain injury.23
Randomisation
Patients are randomised 1:1 in variable block sizes and stratified by centre (prehospital base or trauma centre) and tracheal intubation at inclusion. The randomisation table is generated outside of REDCap by a statistician otherwise not affiliated with the study. The allocation sequence list and block size are only known by the statistician and will remain concealed from the investigators. Prior to randomisation, a proxy consent must be obtained (or according to legislation in each country). Afterwards, randomisation is determined by opening a concealed envelope with information on allocation. The envelopes are available both prehospital and in-hospital. Each concealed envelope contains a study ID that matches a study ID on the randomisation list generated by the statistician.
Participant timeline
Please see figure 1.
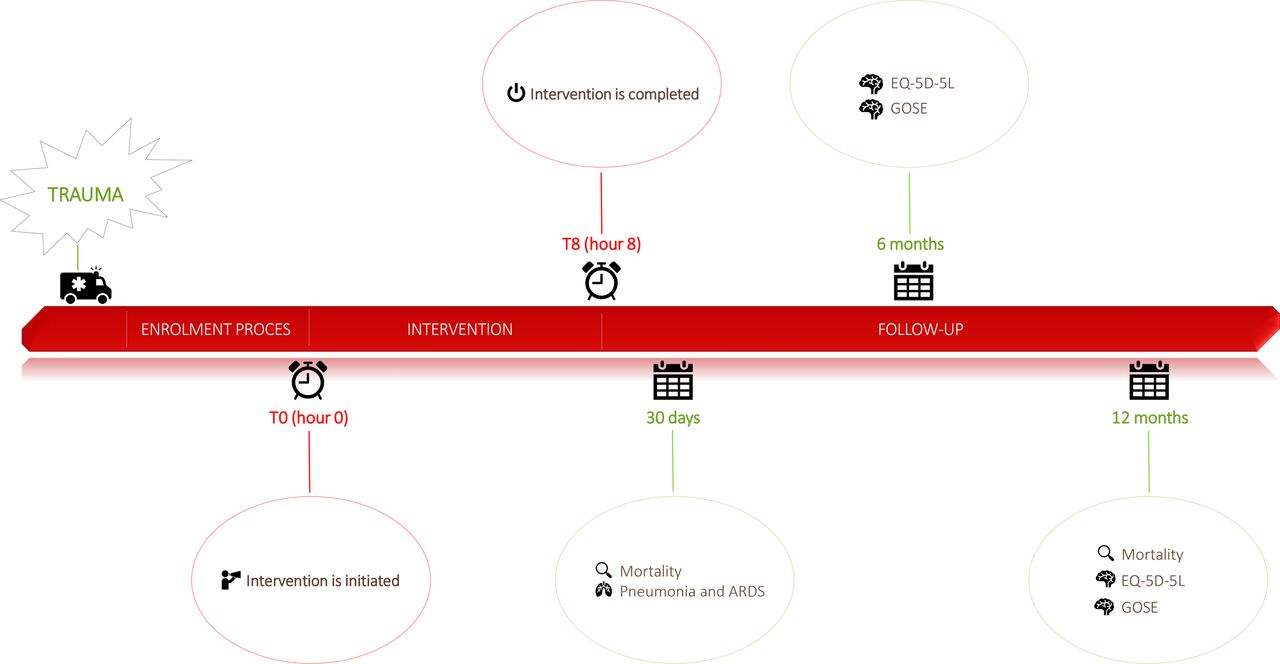
Timeline of patients randomised to either liberal or restrictive oxygen approach from trauma to end of follow-up. GOSE, Glasgow Outcome Scale-Extended.
Blinding
The study outcome assessor-blinded and analyst-blinded with regard to treatment: treating staff are aware of the trial participants’ randomisation group. However, at least two allocation blinded primary outcome assessors (specialists in anaesthesia, intensive care, emergency medicine or similar) are appointed in each country to assess in-hospital lung complications (pneumonia and/or ARDS). Blinding is ensured by concealing all information indicative of the allocation prior to assessment.
The statistician and manuscript writers will be blinded towards the allocation of treatment once the trial ends when data are being analysed and the manuscript is drafted.
Recruitment and assignment of interventions
Depending on the possibilities of the recruiting site, patients are randomised and included either in the prehospital phase or in the trauma bay.
Prehospital phase (optional)
As soon as possible and after immediate and necessary life-saving procedures have been performed, a prehospital emergency physician (the including physician) assesses a patient eligible for inclusion. If all inclusion criteria are met, the including physician obtains proxy consent through a legally appointed study guardian (physician) by telephone. Proxy consent before inclusion may not be necessary in all participating countries. National rules and laws are followed in the specific country on both proxy consent as well as trial participant/trial participant’s next-of-kin consent in general. Once the trauma patient is included, the including physician randomises the trial participant to the intervention (restrictive oxygen strategy) or control (liberal oxygen strategy) group by opening a concealed envelope with information on the allocation. The including physician registers the trial participant in an electronic database. If the trial participant is transported by helicopter, it is advised to fly at the lowest reasonable altitude to reduce the alterations from normal atmospheric oxygen tension at sea level. Treatment is initiated and continued for eight hours after enrolment. Time of initiation equals T0 (see figure 1).
Trauma bay
As soon as possible or after immediate and necessary life-saving procedures have been performed the including physician (eg, the trauma leader or an attending anaesthesiologist) judges whether the patient is eligible for the study. If eligible, the including physician obtains proxy consent through a legally appointed study guardian (physician). Proxy consent before inclusion may not be necessary in all participating countries. National law is followed in the specific country on both proxy consent as well as trial participant/trial participant’s next-of-kin consent in general. Once the trauma patient is included, the including physician randomises the trial participant to the intervention (restrictive oxygen strategy) or control (liberal oxygen strategy) group by opening a concealed envelope with information on allocation. The including physician registers the trial participant in an electronic database. Treatment is initiated and continued for eight hours after enrolment. Time of initiation equals T0 (see figure 1) and must not be delayed more than 90 min after hospital arrival.
Consent
Consent procedures vary according to including location. In broad terms, the patients eligible for inclusion in the trial are temporarily incompetent because of the acute and severe condition related to their traumatic injuries. The trauma patients are eligible either on the scene of accident or on arrival in the trauma bay where early resuscitation with the use of multiple interventions and even surgery may be necessary. Symptoms include severe pain, impaired consciousness and early complications are respiratory and circulatory failure requiring emergency intubation. Some of these trial participants are expected to die. The intervention tested in this trial is pivotal to be given immediately in the early phase of resuscitation. In clinical trials aiming to improve treatment of traumatic injuries, it is necessary to include unconscious and incompetent patients as no clinically relevant animal model exists. As soon as possible, local consent procedures are followed. In Denmark, proxy consent is first obtained through a legally appointed study guardian. Hereafter, consent is sought by the trial participants’ next-of-kin as soon as-possible, and when possible, consent is sought by the trial participant. If the trial participant is not able to consent within 30 days, consent from the next-of kin is accepted as the final consent. If the next-of-kin is unavailable, a secondary proxy consent can be obtained through a legally appointed study guardian, and this can also be accepted as the final consent. Detailed consent procedures in specific locations can be found on the trial website.
Data collection methods, registration, and monitoring
Oxygen dosages and saturations are recorded every hour and two arterial blood gas analyses are obtained and noted in a paper data collection sheet specifically made for this study (‘Randomisation, data collection sheet and REDCap inclusion’, available on the study website). Further data collection is obtained by accessing the trial participants’ medical records. Data points include trial participant characteristics (name, unique patient identifier, age, sex, height and weight), prehospital data (vital signs, trauma mechanism, details on supplemental oxygen in the prehospital phase (indication, SpO2, supplemental oxygen yes/no, intubation yes/no, oxygen flow/FiO2), Injury Severity Score, complete list of injuries, transportation mode to the trauma bay), time points (date and time of trauma, on-scene arrival and departure, trauma bay arrival, ICU/ward arrival, time of intubation/extubation/reintubation, time of surgery, and duration of surgery), HOS LOS and ICU LOS, vital signs on arrival at the trauma bay (including arterial blood gas analysis if available), in-hospital variables (pneumonia, ARDS, other infections (surgical site infection or sepsis)), ischaemic events within seven days after admission (myocardial infarction or cerebral ischaemia), Adverse Events (AE) and Serious Adverse Events (SAE), comorbidities prior to trauma (categorised in heart disease, lung disease and other diseases), active smoker (yes/no), specifics of possible brain injury (type and extent) and other cerebral complications, and time until of death.
Trial participants with traumatic brain injury admitted to a neurosurgical ICU can be monitored according to standard practice in the local facility. It is acceptable, but optional, to perform continuous intraparenchymal brain oxygen measurements.
For all TRAUMOX2 trial participants, it is possible to deviate from the protocol if clinically justified by the treating physician. Such deviations should always be documented including the clinical justification.
Mortality status is collected through local registries or according to local practice. EQ-5D-5L and GOSE score are collected through telephone interviews, either with the trial participant or with the trial participant’s next-of-kin or caregiver. Possible pneumonia postdischarge is evaluated through medicines prescribed after hospital discharge in countries where this information is available.
All trial participants will have two arterial blood gasses (ABGs) drawn within the intervention period. The first ABG is drawn at hour 1±30 min (T1) after randomisation (initiation of intervention is considered hour 0). If an ABG is not obtainable at hour 1 due to eg the patient still being prehospital, the ABG must be obtained as soon as possible. The second ABG is drawn at hour 6±2 hours (T6). If more than two ABGs are collected during the intervention (ABGs not related to the study), the ABGs closest to the specified time slots (T1 and T6) should be used for data entry.
Furthermore, the trial participant or his/her next-of-kin is asked for an additional and separate consent form, not directly related to this research project, to have their blood stored in a biobank established for future research. It is optional whether the participating centres contribute to the biobank.
Data are stored in an electronic, web-based, secure, centralised, user-friendly interface using a data collection sheet in REDCap24 specifically made for this trial. This data management system is secure, fully compliant with all regulatory guidelines and includes a complete audit-trail for data entry validation. Trained members of the research team are responsible for data collection and entry into REDCap using local electronic clinical registries. Therefore, the electronic case report form is digital. In case of system malfunction, a paper version of the data collection sheet is available. The REDCap database is set up from Rigshospitalet in the Capital Region in Denmark and participating centres are invited to enter data in this database.
External monitoring of registered data is applied at all trial centres.
Definitions
Pneumonia is defined as per the CDC criteria.25
ARDS is defined as per the Berlin definition.26
Traumatic brain injury is defined as follows:27
Severe: Abbreviated Injury Scale (AIS)≥5
Moderate: AIS 3–4.
Mild: AIS 1–2.
Please see figure 2 for a detailed description of pneumonia and ARDS.

{kind=link}
{kind=link}
Pneumonia and ARDS (acute respiratory distress syndrome) definitions.
Statistical analysis
In larger studies, mortality from trauma has been estimated to be around 6%–12%,28 and the incidence of ventilator associated pneumonia post-trauma to be almost 30%.14 With 710 trial participants in each arm, we will be able to detect a 33% risk reduction with a restrictive supplemental oxygen strategy (with 80% power at the 5% significance level) if the incidence of our primary outcome is 15% in the liberal group. Our primary analysis will be a modified intention-to-treat analysis, but a per-protocol analysis will also be carried out. A detailed statistical analysis plan, including the prespecified subgroup analyses, will be made before the last patient is included. In the primary analysis, we will exclude trial participants where no injuries are found, defined as Injury Severity Score=0.
If less than 5% of data required for any specific analysis on primary or secondary outcomes are missing, a complete case analysis will be performed. If more than 5% are missing, and it is concluded that data are not ‘missing completely at random’ and inverse probability weighting will be used to correct possible bias.29 A sensitivity analysis on the assumptions used for missing data will be done to verify robustness.
Inclusion of trial participants will end when the goal of 1420 evaluable trial participants has been reached including the 30-day follow-up period. This means that the maximum number of trial participants will be 1600 as inclusion will continue during evaluation of the 30-day follow-up. EQ-5D-5L score and GOSE score at six and 12 months will be obtained. Mortality at 30 days and 12 months will also be obtained. The primary composite outcome will be compared between the two groups using logistic regression reported as OR with 95% CI. The primary analysis will be adjusted for age, sex, centre, intubated at randomisation (yes/no) and known pneumonia on admission (under treatment). Secondary outcomes will also be compared between the two groups using logistic regression for dichotomous data and linear regression for continuously valued outcomes. We will use a 5% significance level. Any changes or additional analyses will be reported.
In the per-protocol analysis, all trial participants with ≥1 major protocol violation will be removed.
Specified subgroup analyses will be made on trial participants initially intubated (within one hour of the accident) (yes/no), trial participants with ICU admission (yes/no), trial participants with moderate and severe traumatic brain injuries (yes/no), trial participants with chronic pulmonary disease (yes/no), registered episodes with hypoxaemia as well as an analysis on trial participants enrolled prehospital versus in-hospital. An analysis adjusted for Injury Severity Score will also be conducted.
The statistical analysis will be performed by a statistician.
Recruitment status
On 19 August 2022, the trial had included 479 patients (34%) and recruitment is thus ongoing.
Adverse and serious adverse events
To monitor adverse events, a TRAUMOX2 investigator assesses the trial participant’s medical record once within the first 24 hours and every third day until discharge (maximum of 30 days).
This group of trial participants are expected to have a lot of complications. It is the established practice in trials on critically ill patients that adverse events are part of the natural trajectory of the primary disease process or expected complications of the critical illness.30 Therefore, we have chosen to record only the occurrence of atelectasis and irritability of airway mucosa.
All SAEs are registered. The registration is done in REDCap and once an SAE registration is completed, the sponsor and coordinating investigator receives an e-mail notification immediately via the REDCap notification e-mail system.
Ethics and dissemination
Trial participant insurances are in place at all trial sites either through the national health insurance or through specifically supplied local trial insurances as required according to the specific trial sites and national regulations.
This RCT is carried out in accordance with the principles from the Helsinki II Declaration in its latest version.31 The protocol has been approved by the Danish Committee on Health Research Ethics for the Capital Region of Denmark (H-21018062) and The Danish Medicines Agency, as well as the Dutch Medical Research Ethics Committee Erasmus MS (NL79921.078.21 and MEC-2021-0932). It is monitored by the regional Good Clinical Practice Unit. Data management must be approved according to national legislation. Furthermore, the trial has been registered in the European Union Drug Regulating Authorities Clinical Trials Database (EudraCT) (2021-000556-19) as well as on www.clinicaltrials.gov (NCT05146700).
Finally, a website (www.traumox2.org) is available for further information and updates on the trial.
Protocol changes
Protocol changes can only be decided by the steering committee. All trial documents, including protocol amendments are available on the public TRAUMOX2 website and communicated to relevant parties when found appropriate.
Timeline of study progress
Inclusion began on 7 December 2021 and is expected to be completed early 2024. Data analysis and manuscript drafting will commence in autumn 2024.,The submission of the primary paper is expected at the beginning of 2025.
Data monitoring and safety committee
An independent data monitoring and safety committee (DMSC) has been set up. The committee includes a statistician. The committee will meet when information on 30-day mortality has been collected in 355 (approximately 25% of the sample size estimation) and 710 (approximately 50% of the sample size estimation) trial participants. Prior to the meeting, a statistician will perform an interim analysis with blinded data provided by the sponsor and principal investigator. Criteria for premature termination will be decided by the steering committee. Furthermore, the sponsor has the responsibility to report the overall number of SAEs yearly to the DMSC. Detailed information on the DMSC is available on the study website.
Publication policy
The study results will be published in an appropriate international peer-reviewed scientific journal. Once the study results are published, it will be announced on the study website. The study is registered and study results will be disclosed by the coordinating principal investigator in one or more public clinical study registry(ies), according to national/international use, including both positive, negative, and inconclusive results. The registration will include a list of the investigational centres. The steering committee and the primary centre investigator (active) will be listed as coauthors in the publications. If the centre involves prehospital inclusion, the prehospital centre investigator (active) will be coauthor. Top-enrolling centres will be able to designate one additional coauthor for every completely documented 100 trial participants. Blinded outcome assessors and the statistician doing the analyses may also qualify as coauthors. All authors must fulfil the criteria for authorship according to the ICMJE group. Each contributing centre can designate a reasonable number of active collaborators that participates in the study administration. These collaborators will be mentioned in the TRAUMOX2-study group and will be trackable via PubMed. In line with the principles of data preservation and sharing, the steering committee will, after publication of the overall data set, consider all reasonable requests to make the data set available in whole or part for secondary analyses and scientific publication. The steering committee will consider additional proposals for secondary analyses based on the scientific quality of the proposal. Proposals will need to be revised and approved by the steering committee prior to submission.
Archiving of documents
The investigator will keep the subject’s files and original data according to the local methods and facilities. The investigator will maintain the trial documents as specified in the ICH-GCP-Guideline for ten years. The investigator/institution will take measures to prevent accidental or premature destruction of these documents.
Patient and public involvement
Patients were not involved in the planning of this study. Trial participants will be given the opportunity to access the outcomes of the study once published.
Discussion
Oxygen has been used for centuries, but the evidence supporting its use remains sparse. The trauma population is particularly exposed to high concentrations of oxygen,1 3 4 10 32 33 although evidence for supplementary oxygen for the trauma population is extremely limited.6 34 This is also evident in the newly updated guidelines on oxygen use in adults in the emergency setting published by the British Thoracic Society.35 In trauma, they recommend initial management with high-concentration oxygen therapy and a target SpO2 of 94%–98% for both hypoxaemic patients and patients ‘at risk of hypoxaemia’. This, however, is a Grade D recommendation.
As mentioned in the introduction, a recently published meta-analysis in the Lancet concluded that clinicians should aim for a target SpO2 of 94%–96% in acutely ill patients.8 Their analysis compared liberal versus conservative oxygen strategies and found increased rates of mortality for patients with SpO2 above 96% compared with 94%–96%. Of note, the trial sequential analysis in the meta-analysis was driven primarily by a single large randomised trial,36 preventing the authors from excluding a small beneficial effect of liberal oxygen therapy. Furthermore, only one study on trauma patients was included.
Nonetheless, there is an increasing concern regarding the detrimental effects of hyperoxaemia, and thus targeting normoxaemia becomes appealing. However, the impact of pursuing normoxaemia on the prevalence of hypoxic episodes is unknown. Normoxia thus suddenly represents the fragile middle ground between two states of which we know one to be harmful and fear the other is too. However, in TRAUMOX1, maintenance of normoxaemia post-trauma appeared feasible and there were few episodes of hypoxaemia.20 Thus, TRAUMOX1 forms the basis of the current trial, TRAUMOX2, that aims to provide high-level evidence on the implications of supplemental oxygen.
In TRAUMOX2, the intervention is an SpO2 target in the restrictive group and an FiO2 target in the liberal group. Given the sigmoid shape of the oxygen dissociation curve, however, the PaO2 resulting from a given FiO2 and SpO2 can vary greatly. Nevertheless, a trial must be feasible, and in the acute phase post trauma, careful titration of the PaO2 is not feasible. A large retrospective study on 864 340 trauma patients found that trauma patients with an SpO2>97% in the emergency department had a higher risk of in-hospital mortality if they received supplemental oxygen.7 A large randomised study on patients with myocardial infarction showed that targeting an SpO2 of 94% resulted in a decrease in myocardial injury and myocardial infarct size.37 Another study has shown a dramatic increase in the occurrence of hyperoxaemia when SpO2 was above 95%,38 and for those reasons, we have chosen SpO2 94% to be the target in the restrictive group. The SpO2 is recorded once every hour during the intervention and aims to represent the median SpO2 during that hour. If multiple measurements are available, for example, in the ICU, the median is calculated. It could be argued that continuous measurements, for example, an SpO2 every minute, would be favourable. However, the trial aims to be pragmatic, and everyday care in the general ward does not allow for careful titration of the SpO2 more than once an hour.
Furthermore, the FiO2 target in the liberal group is based on a guideline where oxygen is recommended for all trauma patients.1 However, the recommendation is without a specific therapeutic goal. In TRAUMOX1, however, some clinicians were concerned about the concentration and duration of oxygen in the liberal group. Therefore, the concentration may be diminished to FiO2 0.60 or 12 L O2/min on a non-rebreather mask once the trial participant reaches the OR, ICU, postanaesthesia care unit, or ward if the saturation is at least 98%.
In a retrospective study of intubated trauma patients, we found that the FiO2 seemed to be high in the first hours after trauma followed by a steady decline until a stable plateau of approximately 0.30–0.40 after 10–12 hours was reached.39 Furthermore, in a randomised trial, administration of 0.80 compared with 0.30 oxygen in the perioperative period (median time with intervention=5.5 hours) was associated with significantly increased long-term mortality.40 Therefore, the duration of the intervention altogether has been diminished from 24 hours to eight hours to ensure representation of only the most acute phase post-trauma until careful oxygen titration becomes possible.
It should be acknowledged that oxygen delivered to the tissues is dependent on numerous factors which are not directly accounted for in this trial, such as cardiac output and haemoglobin levels. In addition, some patients may have pulmonal dysfunction with impaired oxygenation, for instance after chest trauma. The data collected in our pragmatic study will not allow a detailed analysis of all these factors, but haemoglobin levels and the presence of chest trauma are registered. The impact of both could subsequently be explored.
In TRAUMOX1, the median time from trauma to arrival in the trauma bay was 51 (29–68) min. In larger, more geographically challenging countries, however, this time gap may be much longer, and therefore prehospital inclusion is aimed for whenever possible in TRAUMOX2 to diminish time in the acute phase without any allocated intervention begun.
The primary outcome of the trial is the incidence of pulmonary complications and/or death within 30 days (combined outcome). This is done to increase the event rate, but also because both outcomes are very important to the trauma patient: the majority of trauma patients are free from comorbidities and independent prior to their trauma,41 42 but still each year 5.8 million people die as a result of trauma.43 Furthermore, the incidence of pneumonia in trauma patients has been reported to be as high as 26%–44% leading to disability and prolonged hospital stay.44 45 Finally, trauma constitutes a major economic burden, as trauma-related costs were estimated to $671 billion in the USA alone in 2013.46 Understanding whether supplemental oxygen plays a role in the outcome for the trauma patient is thus of utmost importance.
Data availability statement
No data are available. NA.
Ethics statements
Patient consent for publication
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors JB, TA, VS, DLI, LSR and JS: substantial contributions to the conception and design of the work as well as the acquisition, analysis and interpretation of data for the work; drafting the work; final approval of the version to be published; agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. JH, SY, MK, MVV, EVL, SM, STZ, MAS and CFE: substantial contributions to the conception, analysis and interpretation of data for the work; revising it critically for important intellectual content; final approval of the version to be published; agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding TRAUMOX2 is initiated by a research group of medical doctors in 'Department of Anaesthesia and Trauma Centre, Centre of Head and Orthopaedics, Rigshospitalet, Denmark'. The work presented in this article is supported by Novo Nordisk Foundation grant NNF20OC0063985. In addition, the study is supported by The Lundbeck Foundation through a personal grant to author JB. Drug expenses are covered by the participating centres’ departmental budget. Drug expenses beyond the normal department budget is also be covered by the participating centres.
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.