Article Text

Abstract
Introduction Clinically complex patients often require multiple medications. Polypharmacy is associated with inappropriate prescriptions, which may lead to negative outcomes. Few effective tools are available to help physicians optimise patient medication. This study assesses whether an electronic medication management support system (eMMa) reduces hospitalisation and mortality and improves prescription quality/safety in patients with polypharmacy.
Methods and analysis Planned design: pragmatic, parallel cluster-randomised controlled trial; general practices as randomisation unit; patients as analysis unit. As practice recruitment was poor, we included additional data to our primary endpoint analysis for practices and quarters from October 2017 to March 2021. Since randomisation was performed in waves, final study design corresponds to a stepped-wedge design with open cohort and step-length of one quarter. Scope: general practices, Westphalia-Lippe (Germany), caring for BARMER health fund-covered patients. Population: patients (≥18 years) with polypharmacy (≥5 prescriptions). Sample size: initially, 32 patients from each of 539 practices were required for each study arm (17 200 patients/arm), but only 688 practices were randomised after 2 years of recruitment. Design change ensures that 80% power is nonetheless achieved. Intervention: complex intervention eMMa. Follow-up: at least five quarters/cluster (practice). recruitment: practices recruited/randomised at different times; after follow-up, control group practices may access eMMa. Outcomes: primary endpoint is all-cause mortality and hospitalisation; secondary endpoints are number of potentially inappropriate medications, cause-specific hospitalisation preceded by high-risk prescribing and medication underuse. Statistical analysis: primary and secondary outcomes are measured quarterly at patient level. A generalised linear mixed-effect model and repeated patient measurements are used to consider patient clusters within practices. Time and intervention group are considered fixed factors; variation between practices and patients is fitted as random effects. Intention-to-treat principle is used to analyse primary and key secondary endpoints.
Ethics and dissemination Trial approved by Ethics Commission of North-Rhine Medical Association. Results will be disseminated through workshops, peer-reviewed publications, local and international conferences.
Trial registration NCT03430336. ClinicalTrials.gov (https://clinicaltrials.gov/ct2/show/NCT03430336).
- general medicine (see internal medicine)
- health & safety
- change management
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
We will provide evidence of the effectiveness of an electronic medication management support system in reducing mortality and hospitalisation in adult patients with polypharmacy in real-life general practice.
The intervention concept is innovative, as it is the first time that information based on claims data is made available to general practitioners (in Germany) in the form of an electronic tool.
However, claims-based outcome measures also have disadvantages, as data are collected for the purpose of reimbursement, which limits the choice of outcomes.
A stepped-wedge cluster-randomised design with an open cohort will allow us to overcome insufficient recruitment.
We included a time variable to adjust for confounding time effects and overcome such methodological shortcomings of stepped-wedge design.
Introduction
Multiple medications are often required to manage clinically complex patients. Clinicians are frequently challenged by the need to ensure that treatment of complex patients adheres to disease-specific clinical practice guidelines.
Polypharmacy, defined as the use of five or more medications,1 increases the potential for the prescription of potentially inappropriate medications (PIMs) due to the non-consideration of drug–drug or drug–disease interactions, inappropriate dosages (perhaps due to the age of the patient) as well as unintended duplicate prescriptions.2–6 The use of greater numbers of drug therapies has been associated with increased risk of adverse drug reactions7 irrespective of age.8 It has also been associated with increased risk of hospital admissions,9–11 hip fractures in older adults12 and higher costs and mortality.10 11 13
In line with the increasing number and complexity of medications, polypharmacy is associated with reduced medication adherence in patients. It may also result in undertreatment, particularly in the elderly, in whom too few prescriptions and excessively low dosages have been reported.14–16
Medication errors and omissions are important problems facing routine care in general practice, especially in patients with multimorbidity and multiple prescriptions.17–19 They may contribute to patient hospital admissions and mortality, thus additional understanding of such incidents is required.20 As most medication errors and omissions are preventable, raising physicians’ awareness of polypharmacy may help to ensure the safe, effective and appropriate use of medication.19 21 22
Medication management strategies allow patients and families to actively participate with their physicians in developing complete and accurate medication lists. To ensure patients receive high-quality healthcare, physicians should be provided with tools that help them avoid risks in the treatment of their patients.22–24 Likewise, physicians should have access to continuously available data on quality-oriented aspects to support the control of their patients’ treatments.24 Few effective instruments are available to help physicians systematically monitor and optimise the medications their patients take.22 Such tools comprise computerised Decision Support Systems (CDSS) or complex multifaceted pharmaceutical care-based approaches that may incorporate CDSS as part of the intervention. CDSS are computer-based systems providing ‘passive and active referential information as well as reminders, alerts and guidelines’.25 A recent systematic review26 concluded that although CDSS may reduce PIMs, additional randomised controlled trials are needed to assess their impact on patient-relevant outcomes and to evaluate the use of medication targets such as the Screening Tool of Older People’s Prescriptions and the Screening Tool to Alert doctors to the Right Treatment (START) criteria.27
Considering that individual, patient-related information relevant for the drug therapy is currently unavailable to physicians and that there is a lack of instruments helping physicians to regularly review their patients’ medication, an intervention with a web-based medication management system was developed within the Anwendung für digital unterstütztes Arzneimitteltherapie-Management (AdAM) project. The primary objective of the AdAM trial is, therefore, to assess whether such electronic medication management support system (complex intervention) reduces the combined endpoint of all-cause mortality and all-cause hospital admissions in patients with polypharmacy, compared with usual care and in the real context of a general practice setting. Substudies to be performed will include cost-effectiveness analysis, the analysis of barriers and facilitators through interviews and focus groups with practitioners and interviews with patients, a trial process evaluation as well as sustainability analysis and quality cost accounting systems to explore the relationship between organisational context, implementation process and quality of care (online supplemental additional file 1). However, as this study protocol focuses on the AdAM intervention, these substudies will not be explained in detail in this paper.
Supplemental material
Aims
The AdAM trial aims to:
Evaluate whether the complex intervention reduces the combined outcome of all-cause hospitalisation (including night-only and day-only admissions) and all-cause mortality (primary outcome) or any of its components (secondary outcomes) in patients with polypharmacy, compared with usual care.
Evaluate whether the complex intervention reduces cause-specific hospitalisation preceded by high-risk prescribing in patients with polypharmacy, compared with usual care (secondary outcomes).
Ascertain whether the complex intervention reduces the number of PIMs and Potential Prescribing Omissions as measured using explicit criteria, in patients with polypharmacy, compared with usual care (outcomes of process of care).
Assess whether the complex intervention reduces the number of prescribed medications in patients with polypharmacy, compared with usual care (outcomes of process of care).
Evaluate whether the complex intervention is effective in reducing the combined primary outcome, or any of its components, in subgroups of patients defined according to age (<65 vs ≥65 years), sex, early and late enrolment (patient does or does not fulfil the inclusion criteria from the moment he or she joins the intervention of the associated practice) and main treating physician (general practitioner—GP vs specialised physician or hospital outpatient clinics).
Methods and analysis
Study design
The AdAM trial was originally planned as a pragmatic, parallel cluster-randomised controlled trial (cRCT) with 15 months (five quarters) of follow-up per cluster (practice). The general practice was the unit of randomisation and the patient the unit of analysis. Since GPs trained in performing the intervention are unable to provide usual care, a clustered design (practices as clusters) was chosen to reduce treatment group contamination.
Important changes after trial launch
When practice recruitment ended in June 2019, it became obvious that the target numbers of practices and patients would not be achieved. Extensive simulations were, therefore, conducted on the assumptions that the number of eligible patients was the same (39 per practice) in all 688 randomised practices, that 60% of potential patients had enrolled and that the event rate in the control group would be constant in all quarters. After completing the simulation, we decided to change the design of the trial in such a way that a power of 80% could still be reached. The following changes were made and will be explained in detail in each section of the protocol: (1) primary and secondary outcomes will be measured at regular intervals over 12 quarters, rather than once after five quarters and (2) The statistical analysis will be adapted to take account of the new design.
All changes were made before data from the study population were analysed (figure 1).
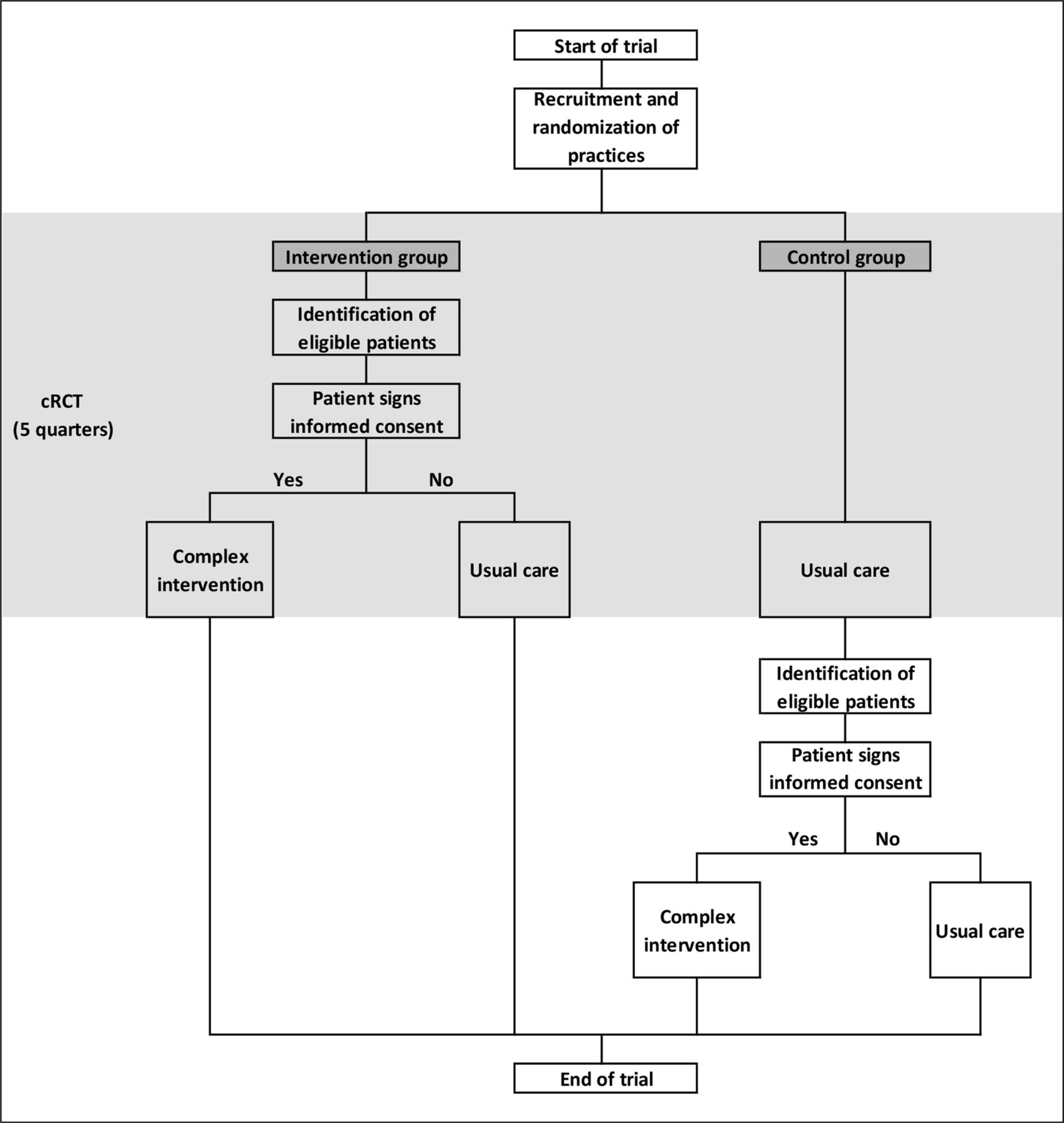
AdAM study flowchart. AdAM, Anwendung für digital unterstütztes Arzneimitteltherapie-Management; cRCT, cluster-randomised controlled trial.
Study setting and population
The trial is conducted in general practices in Westphalia-Lippe, Germany.
Inclusion criteria for trial sites (general practices)
All criteria had to be fulfilled:
General practices provide health services to patients covered by the BARMER statutory health insurance fund (BARMER).
Physicians work as GPs and have specialised in general practice, internal medicine or in no particular field.
Practices have at least 10 eligible patients.
Practices have access to the Westphalia-Lippe Association of Statutory Health Insurance Physicians (KVWL) website through a secure connection that can be used by both GPs and other medical staff (practice nurse and healthcare assistants).
Investigators agree to fulfil the contractual obligations arising from the trial.
Inclusion criteria for patients
All criteria had to be fulfilled:
Patients are at least 18 years of age and covered by BARMER.
They have polypharmacy, defined as the regular intake of at least five drugs (≥ five different Anatomical Therapeutic Chemicals—ATC) in at least one quarter of the previous year. Each of the five ATCs has to be prescribed over at least two consecutive quarter in the previous year.
In order to participate in the intervention, patients had to provide written informed consent (online supplemental additional file 2). They also had to be competent to sign the required documents under law and capable of providing written informed consent to participate in the trial voluntarily. Patients who were not competent to sign the documents under law and were not capable of providing written informed consent to participate in the trial voluntarily (eg, because of dementia) could provide written informed consent signed by an informal caregiver.
Supplemental material
No changes were made to setting and study population after trial launch
Recruitment and registration
Recruitment and registration of practices
The KVWL and the BARMER provided a list of general practices that were eligible to participate in the trial. Of these, the KVWL contacted GPs from practices with at least 10 eligible patients by postal mail (written invitation). Reminders were later sent by fax. GPs who wished to participate had to return a signed investigator’s agreement form to the KVWL (either by postal mail or fax).
Moreover, the trial was announced in journals and local media (press, radio, television) and communicated to local key stakeholders (moderators of quality circles, managers of practice networks, etc). Local recruitment events were also organised.
Recruitment and registration of patients
STEP 1: before randomisation and quarterly during the intervention period, the BARMER identified eligible patients from the participating general practices based on claims data.
STEP 2: after cluster-randomisation of participating practices, patients in the intervention practices were recruited in three ways:
Every quarter, GPs received a list of eligible patients as well as written information and informed consent forms for the patients. The GPs could, therefore, invite eligible patients on their lists to participate.
The BARMER sent written information on the study (information letter and a flyer) to eligible patients from participating intervention practices, so that they could actively approach their GPs to find out about the study. The aim was to explain the contents of the AdAM project to eligible patients in good time in order to arouse interest and actively assist in enrolment. The BARMER telephone hotline was available to immediately answer any questions the patients had. Additional information on the study was provided on the BARMER website (daily news and Frequently Asked Questions (FAQ) list).
GPs invited patients from their practices that fulfilled the inclusion criteria but had not (yet) been identified as eligible from claims data (eg, due to a delay of data processing).
STEP 3: GPs sent patients’ written informed consent to the KVWL. The KVWL digitised the consent forms and transmitted them to BARMER for verification of insurance status. When the results were positive, KVWL permitted GPs to access the electronic medication management support system (eMMa) and forwarded the original consent forms to the BARMER for archiving.
When the follow-up period of the cRCT was over, eligible patients in the control group that were identified in STEP 1 were invited to provide their written informed consent and participate in the intervention. Beginning with STEP 2, the recruitment and registration of control patients followed the same procedure as intervention patients (figure 1).
No changes were made in recruitment and registration after the trial began
Randomisation and allocation concealment
Practices were randomly allocated to the complex intervention or control arm in a ratio of 1:1 (figure 2). Balanced randomisation was performed every month to ensure that the treatment groups were of approximately equal size for each quarter. The KVWL provided lists of participating practices to the Department of Medical Informatics, Biometry and Epidemiology (AMIB) at the Ruhr University Bochum, Germany. A study-independent staff member at the AMIB used computer-generated random numbers to generate randomisation lists from the list of participating practices. Randomisation lists were sent to KVWL, which concealed treatment allocation to participating practices. Once a practice was randomised, all eligible patients at the practice were deemed to be intervention or control patients, depending on the arm of the study the practice was allocated to. The first list of eligible patients in the intervention group was made available to participating physicians and the intervention began, after patients had signed the informed consent form. Eligible patients in the control group continued to receive usual care. After signing the informed consent form, eligible patients in the control group were invited to participate in the intervention five quarters after the start of the intervention at the other practices from the same randomisation wave.

{kind=link}
{kind=link}
AdAM data availability (time flow). AdAM, Anwendung für digital unterstütztes Arzneimitteltherapie-Management; cRCT, cluster-randomised controlled trial.
No changes were made in randomisation and allocation concealment after the trial began
Blinding
Allocation was disclosed to the practices soon after randomisation and to patients from intervention practices when they were asked to provide their written informed consent. Patients in the control group were not aware of the study until the end of their practice’s follow-up period of the cRCT.
Due to the type of intervention, neither GPs and their patients nor the AdAM study team was blinded to the treatment allocation.
No changes were made in blinding after trial commencement
Treatment plan for intervention and control groups
Intervention group
Several key elements of the intervention must be put into place in participating general practices:
The web-based, user-initiated CDSS eMMa provides the GP with drug–therapy information that is relevant to participating patients with polypharmacy on demand. The information might include data on diagnoses, treatments (also non-pharmacologic, such as physiotherapy) and medical products (eg, assistive devices). The information is based on claims data gathered from all healthcare professionals involved in the care of the patient (eg, specialised ambulatory care physicians, other GPs, psychotherapists as well as data on hospital stays and prescription data from pharmacies). RpDoc Solutions GmbH developed eMMa in collaboration with KVWL.
GPs can add and modify patient data in eMMa (eg, remove drugs that the patient no longer takes, add new diagnoses, prescriptions and over the counter (OTC) drugs and recent laboratory findings about kidney function, etc) in order to enhance and update relevant information.
Aided by eMMa, GPs systematically assess the appropriateness of every patient’s medication at least once a year. Alerts will draw the GP’s attention to possible drug–drug interactions, drug–disease interactions, age-related PIMs, duplicate medications, renal dose adjustments, allergies, as well as general inappropriateness, such as prescriptions associated with Dear Doctor letters (Rote-Hand-Briefe) and QT prolongation (for a detailed description see online supplemental additional file 3).
GPs optimise patient medication.
GPs print out the updated medication plan, which includes recommendations on medication use, reasons for prescriptions in lay language, and information on drugs that should be avoided, and hand it out to patients. The plan will also be available in foreign languages for patients that speak poor German.
eMMa provides GPs with guidance (eg, recommendations addressing certain types of medication errors and high-risk prescribing that were developed by the German Society for Internal Medicine in collaboration with other scientific medical societies).
Supplemental material
Intervention training
GPs were invited to attend two kick-off meetings and a decentralised event on polypharmacy with a consulting pharmacist from KVWL.
GPs and healthcare assistants also could attend a decentralised software training event with consulting pharmacists and IT support staff.
The KVWL has made a training video and an FAQ list for participating practices available on the trial access site.
During practice hours, several telephone hotlines were offered for technical questions (IT support) and to provide on-site support for questions relating to administration, management and use.
The Template for Intervention Description and Replication (TIDieR) checklist was used to ensure that the intervention reporting standards were met (online supplemental additional file 4).
Supplemental material
No changes were made to the experimental treatment after the trial commenced.
Control group
For the duration of the cRCT, patients in the control group continued to receive usual treatment from their GP. Five quarters after the start of the intervention at the other practices from the same randomisation wave, control practices could switch to intervention and the patients in these practices had the option to switch to the intervention group on condition that they first provide their written informed consent to receive the intervention.
No changes were made concerning the control group, as the switch to the intervention group was already planned in order to carry out the substudy on sustainability (see online supplemental additional file 1).
Outcome assessment
Primary outcome
The primary outcome is the combined endpoint of all-cause mortality and all-cause hospitalisation (including night-only and day-only admissions) in patients with polypharmacy, as assessed quarterly (table 1).
Primary outcome measure—CPO—all-cause mortality and all-cause hospitalisation
Secondary outcomes
1. All-cause hospitalisation (quarterly): to evaluate whether the complex intervention reduces all-cause hospitalisation (including day-only or night-only admissions) (number and duration) in patients with polypharmacy (table 2).
Secondary outcome measures—hospitalisation* (SOh)
2. All-cause mortality (quarterly): to assess whether the complex intervention reduces all-cause mortality in patients with polypharmacy (table 3).
Secondary outcome measure—mortality (SOm)
3. Incidence rate of cause-specific hospitalisation preceded by high-risk prescribing (quarterly): to evaluate whether the complex intervention reduces cause-specific hospital admissions (gastrointestinal bleeding, heart failure, renal failure, fall-related fractures or injuries; including and excluding day-only admissions) preceded by high-risk prescribing in patients with polypharmacy (table 4).
Secondary outcome measures—cause-specific hospital admissions (SOh)
Secondary outcomes concerning process of care
4. Number of PIMs (quarterly): to ascertain whether the complex intervention improves the appropriateness of prescriptions in patients with polypharmacy (tables 5 and 6).
Secondary outcome measures—PIM-related high-risk prescribing (SOpim)
Secondary outcome measures—PIM-related high-risk prescribing composite (SOpim)
5. Total number of underused medications (quarterly): to assess whether the total number of underused medications (based on the modified START criteria) in patients with polypharmacy does not increase in the intervention group in comparison to the control group (table 7).
Secondary outcome measures—underused medication (SOum)
6. Total number of prescribed medications (quarterly): To assess whether the complex intervention reduces the total number of prescribed medications in patients with polypharmacy (table 8).
Secondary outcome measures and process measures—polypharmacy indicators (SOp)
Data for primary and secondary outcomes will be taken from health insurance claims data (BARMER) for the period from the fourth quarter 2017 to the first quarter 2021.
Changes made after trial commencement: initially, we planned a one-time survey of outcomes for a period of five quarters following randomisation. In the end, data on the endpoints were collected quarterly for the period from the fourth quarter 2017 to the first quarter 2021.
See online supplemental additional file 5 for more information about the secondary outcome measures.
Supplemental material
Explanatory variables for population characteristics
Patient (first level) variables
Sociodemographic patient data: sex, age, insurance status and reason insurance coverage ended (death, change of sickness fund).
Outpatient diagnoses and outpatient services: the International Classification of Diseases 10th edition codes28 are used for the outpatient diagnoses, which are documented on a quarterly basis. The services are coded according to the Physician’s Fee Scale (Einheitlicher Bewertungsmaßstab).
Medication: drugs are identified using their national drug code (pharmaceutical registration number, Pharma-Zentral-Nummer—PZN), which contains all relevant information such as trade name, active chemical ingredient(s), strength, application, dosage and indication. The PZN will be linked to the ATC Classification System, which allows analysis to be based on active ingredients, manufacturer and package size. The duration of the therapy will be assessed by means of the defined daily dose and included in the reference table. The data set only includes prescribed medication that is paid for by the insurance fund.
Inpatient data: for each hospitalisation, the start and end date, the admission and discharge diagnosis (with date), as well as secondary diagnoses, will be available. Furthermore, operations and treatment procedures are also documented (Operation and Procedure—Code).
Long-term nursing care (Sozialgesetzbuch): for patients receiving long-term nursing care, the start and end date, the level and place of care, the costs and type of services (cash, non-cash, combined) are documented in the data set.
Practice profile (second level) variables
Single-handed practice/group practice (including ambulatory healthcare centres, along with the number of physicians).
Work experience (start and end date of practice according to KVWL data).
Practice size: number of registered patients in most recent quarter.
Participation in a (regional) practice network.
GP profile (second level) variables
Age, gender.
No changes were made to explanatory variables
Safety monitoring and adverse events
Safety and adverse events were not monitored and reported on, since it was assumed that treatment could not deteriorate as a result of the trial. The study team had no influence on the diagnostic–therapeutic decision-making of GPs and their patients, and analysis of the pseudonymous data will be conducted with a significant delay. GPs and patients could, therefore, not be informed of identified medication errors.
Unintended consequences of using the e-Health technology such as non-acceptance will be investigated qualitatively (online supplemental additional file 1).
Data collection and management
Data collection
Information on all eligible patients was taken pseudonymously from BARMER’s claims data. Claims data detail billable interactions (insurer claims) between the insured patients and the healthcare delivery system.
In the trial, the KVWL data are not systematically linked to BARMER’s data on either a practitioner or patient level. The KVWL provides sociodemographic data on GPs and practice profiles for both the intervention and control groups.
Data management
The required claims data for all eligible patients in the region covered by the KVWL will be specified in a coordinated Minimum Data Set (MDS) and prepared by the PMV research group in Cologne.
The trial data will be archived for 10 years. BARMER will archive a back-up copy containing the data of all study patients (list of eligible patients, declarations of consent to participate in the trial and on data protection, signed and dated by the patients as well as the data provided for the evaluation) in accordance with European basic data protection regulations. The KVWL will archive documents concerning the general practices/GPs participating in the trial (eg, signed investigator’s agreement form). The Institute of General Practice (IGP) will archive the trial master file and any related study plans (MDS and statistical analysis plan). The data provided by KVWL and eMMa, as well as primary data collected in interviews with patients, will be archived by the IGP in accordance with European basic data protection regulations.
End of the trial
The regular end of the intervention and follow-up period for all patients was March 2021.
A patient’s participation in the intervention ends prematurely: (1) when he or she switches to another insurance company and/or a non-participating practice or (2) the GP withdraws his or her consent or is no longer licensed to provide health services by the KVWL.
Schedule and duration of the trial
Practice recruitment: 2 May 2017 to 30 June 2019.
Intervention period: 15 February 2018 to 31 March 2021.
Claims data from 1 January 2017 to 31 March 2021 will be used in the analysis. The cohort is open, meaning that patient data are included from the quarter in which the inclusion criteria are met.
Quality control and quality assurance
The principal investigator and a steering committee (comprising representatives of BARMER, KVWL and the evaluation team) guarantee that all processes in the trial comply with Good Clinical Practice (GCP) guidelines and ethical and legal requirements.
BARMER and the KVWL are responsible for monitoring the trial and were in particular responsible for the recruitment of practices and patients, randomisation (supported by the AMIB), the implementation of the intervention and the provision of data to the evaluation team.
A designated advisory board provides advice on questions concerning planning, conducting and analysing the trial.
Changes to data collection and data management: initially, data collection for each practice was to be carried out as a one-time survey to take place after the start of randomisation and over a period of five quarters. In the end, data were collected at regular intervals over 12 quarters from the fourth quarter 2017 to the first quarter 2021 (light blue and light red areas in figure 2).
Sample size
Initially, based on data detailing the incidence of hospitalisation and all-cause mortality in patients with multiple prescriptions, we expected rates of 30% in the control group over a 12-month follow-up period.16 17 Based on a duration of 15 months (five quarters), the rates were assumed to be 35.25% in the control group, with a relative reduction of 5% in the intervention group. Based on 80% recruitment of practices and patients and an intra-cluster correlation coefficient of 1%, a sample size of 17 200 cluster-randomised patients per group (539 practices per study arm, about 32 patients per practice) is required to detect an absolute difference in the combined endpoint of 1.8% between intervention and control groups (type 1 error of 5% and type 2 error of 15%).
Changes made after trial launch: at the end of practice recruitment in June 2019, it became clear that the target numbers of practices could not be achieved. In the period from 27 June 2017 to 03 July 2019, 688 practices were randomised to the intervention and control groups. Based on the assumptions of 26 832 (688*39) eligible patients in the randomised practices, a participation rate of 60% of patients in the intervention group, the same number of practices at all changeover times (ie, the switch from control to intervention group) and a constant event rate in the control group over all quarters, a power of 80% is achievable.
Statistical analysis
Population for analysis
As both patients who met the inclusion and exclusion criteria from the beginning and patients who fulfilled the inclusion and exclusion criteria after the trial had commenced were able to receive the intervention, the ITT population was an open cohort. Patients from participating practices, therefore, started from the time at which inclusion and exclusion criteria were met during a period stretching from the fourth quarter 2017 to the end of the first quarter 2021. Following the ITT principle, practices and their patients will be analysed quarterly, according to the group to which the practice was allocated, regardless of whether they refused or discontinued the allocated treatment, or whether there were other deviations from the protocol.
For the efficacy analysis, only patients who were selected from the intervention group and for whom the GP had performed the intervention will be considered. This subgroup will be compared with patients in the control group that started the intervention after completion of the cRCT phase. In this population, it will be possible to estimate the maximum possible effect of the intervention, comparable to a per-protocol population.
No changes were made to the population for analysis.
Statistical hypotheses, methods and analyses
The primary objective of this study is to determine whether the complex intervention reduces the combined endpoint of all-cause mortality and all-cause hospitalisation (including night-only and day-only admissions) in adult patients with polypharmacy, as compared with usual care. Statistically, the study objective is formulated as a test of the null hypothesis H0: p1=p2 (the two groups do not differ in terms of the quarterly event probability of combined endpoint pi, where i=1 or 2 for intervention or control group, respectively), compared with the alternative hypothesis H1: p1≠p2 (there is a difference between the two groups).
The analysis is based on quarterly data at a patient level and patients are clustered in practices. We will adjust for the different observation periods and for clustering in the data by fitting an appropriate generalised linear mixed model (GLMM). A mixed logistic regression model will, therefore, be used for all binary outcomes, and especially for the primary endpoint.
Time and treatment group and further confounders such as age, sex, the medCDS prognostic index,29 care level/degree at baseline, days in hospital in the 12 months preceding baseline are considered to be fixed factors. Since all practices were observed under both control and intervention conditions, it will be necessary to include two correlate random cluster-level effects in the model. To gauge the interdependence of individual measurements OTC of the study, additional uncorrelated random effects for patients will also be fitted.
In the AdAM trial, we have assumed that the intervention requires an initial period of adjustment before becoming fully embedded. The intervention effect is, therefore, expected to gradually increase from the time the practice switches to the intervention (¼ in the quarter of the practice change, ½ in the quarter after the change to intervention and the full effect thereafter).
A similar approach will be used to investigate secondary outcomes, sensitivity and efficacy.
The secondary outcomes 2 (all-cause hospitalisation) and 3 (all-cause mortality) are to be analysed hierarchically, reflecting the rationale of the intervention, with a significant decrease in the combined primary endpoint of all-cause mortality and all-cause hospital admissions (level 1) expected to reflect primarily in a decline in all-cause hospitalisation (level 2). If so, all-cause mortality may also decrease (level 3). Therefore, the prespecified secondary outcomes 2 and 3 will be tested in a confirmatory manner. If no significant differences occur at any level, tests of outcomes on higher levels will be exploratory.
The baseline characteristics of participating practices, GPs and patients will be described according to the initially allocated treatment arm. Categorical data will be presented as frequencies and percentages. Total numbers, mean, SD, median, IQR, minimum and maximum will be provided for continuous data.
All statistical tests will be two sided at a significance level of α=0.05. No interim analysis of efficacy will be performed.
Changes made after trial launch: we initially planned to use a GLMM to evaluate the treatment effect in a randomised parallel group design. In addition to considering the treatment group to be a fixed factor, a random effect to account for clustering patients in practices is necessary. Due to the switch to a stepped-wedge design, a more complex model structure was required (see above).
Patient and public involvement
This protocol was developed without patient or public involvement.
Ethics and dissemination
The project is being carried out in accordance with the Medical Association’s code of conduct and GCP, and in line with the World Medical Association Declaration of Helsinki.30 The study plans and all patient-related documents have been sent to and approved by the Ethics Commission of the North-Rhine Medical Association (approval date 26 July 2017, approval number 2017184).
All changes made and reported here after the trial began have also been sent to and approved by the above-mentioned ethics committee (approval date 3 April 2020, approval number 6000207769).
The voluntary participation of practitioners in the trial is recorded in writing following their informed decision. Patients were asked for their consent as soon as the practice switched to the intervention. Patients who did not wish to participate continued to receive usual care.
Data protection is guaranteed for all patient-related data. Eligible patients were identified using pseudonymous claims data from BARMER, whereby BARMER previously informed the patient of the opportunity to participate in the trial. Before the intervention began, patients were separately informed about data protection during the trial and intervention. Patients had to provide their informed consent by signing and dating a declaration.
This study protocol was prepared in accordance with the extension of the Consolidated Standards of Reporting Trials 2010 statement for reporting on cluster randomised trials (online supplemental additional file 6)31 and the Standard Protocol Items: Recommendations for Interventional Trials 2013 statement for reporting on clinical trial protocols (online supplemental additional file 7).32
Supplemental material
Supplemental material
We will prepare presentations to disseminate the study findings to healthcare stakeholders and patients, and at relevant national and international conferences. We aim to publish the results of the trial in peer-reviewed journals.
Ethics statements
Patient consent for publication
Acknowledgments
We would like to thank all practice teams and patients for their participation in the study and also Sandra Rauck and Kiran Chapidi for their contributions as data managers. We appreciate the project management support from BARMER and KVWL. We also thank Phillip Elliott for the language review of the paper.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
BSM and RK-M are joint first authors.
Twitter @rafaoxford, @@PfaffHolger
Collaborators AdAM study group: Julia Jachmich, Eva Leicher, Benjamin Brandt, Johanna Richard, Frank Meyer, Mathias Flume, Thomas Müller, Kiran Chapidi, Robin Brünn, Jale Basten, Karolina Beifuß.
Contributors AIG-G drafted the first version of the manuscript with input from BSM and CM. Critical revision of manuscript for important intellectual content: RK-M, NT and HJT. CM, HJT, FMG, DG, WG, SH, RP, PG, HP, UK, PK-M and PI are responsible for study concept and design. PK-M is the study director. Acquisition of data: BSM, BF, RH, PK-M, TB, LD, TSD, SG, JK-N, AP, UK, SS. Analysis and interpretation of data will be performed by RK-M, JK-N, HJT, BS, PI, SS, UK, AP, WG, CM, NT, IM. PI is responsible for strategic data management. CM and NT are the chief investigators of the study. All authors reviewed the paper and read and approved the final manuscript. CM and NT shared the last authorship.
Funding This study was funded by the Innovation Fund of the German Federal Joint Committee (grant no 01NVF16006).
Disclaimer The funder had no role in the design of the study, or in writing the manuscript.
Competing interests BSM, RK-M, AIG-G, RH, JK-N, PK-M, NT, TB, LD, BS, BF, PI, SS, TSD, AP, IM, UK, HP, WG, FMG, HJT, CM report grants from the German Federal Joint Committee during the conduct of the study. DG reports grants from BARMER during the conduct of the study and family member works for and holds shares of IT company involved in the project. SG works for and holds shares of IT company involved in the project. SH, RP, PG declare that they have no competing interests.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.