Article Text

Statistics from Altmetric.com
In 1896 Antoine Marfan, a French paediatrician, presented a 5 year old girl to the Medical Society of the Paris Hospitals. She had striking abnormalities in her skeletal system with elongation of her long bones and fingers. Association of arachnodactyly with dislocated lenses was described in 1914 and the adoption of this as a syndrome was based upon these characteristic phenotypic appearances. Autosomal dominant inheritance was recognised in 1931. The first descriptions of a dilated aortic root and of dissection were in 1943.
The main cause of premature death in those with Marfan syndrome is dissection of the ascending aorta resulting in tamponade, left ventricular failure caused by aortic regurgitation, myocardial ischaemia from disruption of the coronary orifices, and stroke if the arch vessels are involved. Reports vary but a life expectancy of between 30–40 years of age was typical in the era before root replacement, with most of these premature deaths being attributable to aortic disease.1 ,2 Avoidable deaths in the teens and 20s are not rare. There is a great clinical variability so the pool of patients, and hence the denominator against which the death rate is calculated, depends upon the clinical discipline of those collecting the cases and whether the emphasis is on lethal aortic manifestations, or on abnormalities of the eyes, or on spinal deformity. Neonatal Marfan syndrome has an extremely poor prognosis. Those who have survived to present in mature adult life are a favourable subset of the total spectrum of cases. Different clinics collect rather different sets of people with Marfan syndrome, but for those with an abnormal aorta the expectation of life is greatly improved by aortic root replacement.3 ,4
Diagnosing Marfan syndrome
Diagnosis by nosology
The Berlin Nosology of Heritable Disorders of Connective Tissue was published in 1986.5 Under the headings skeletal, ocular, cardiovascular, pulmonary, skin, and central nervous system it lists four major manifestations—ectopia lentis, aortic dissection, dilatation of the ascending aorta, and dural ectasia—and a host of minor manifestations including arrhythmia and endocarditis. These last items indicate the arbitrariness of a nosology based on clinical features. The Berlin nosology has been replaced by the Gent criteria6 that include the same major cardiovascular manifestations.
Genetic diagnosis
Although Marfan syndrome was clearly recognised as a disorder of connective tissue from the 1950s, the responsible component of the extracellular matrix was variously believed to be collagen, elastin, and hyaluronic acid. In the late 1980s fibrillin abnormalities were reproducibly identified by immunohistochemistry. The gene was found on chromosome 15. It appeared that there would be one absolute criterion for the diagnosis—did the individual carry the abnormal fibrillin gene or not? Unfortunately it turned out to be not nearly so simple. Nearly all the mutations have been family specific. More than 50 causative mutations have been found—nearly as many as families studied—explaining the variability on the clinical manifestations across the spectrum of Marfan syndrome, but not the relative consistency within families. Increasing knowledge of genotype has not allowed prediction of phenotype and therefore risk of aortic dissection for the individual.
We already knew that about 25% of cases have no family history and must be caused by sporadic mutations. It would not be too surprising to find that these individuals are statistically more likely to dissect, for in order to feature in a family tree the gene must be compatible with survival at least into adult life. Thus, even if we think we are being very scientific in making the diagnosis on a genetic or a molecular basis, the diagnostic group includes considerable variability from one case to another, not only clinically7 ,8 but in terms of the ultrastructural and the exact chromosomal abnormality.
Diagnosis by gross morphology
The normal aorta has three gentle bulges, the aortic sinuses, just distal to the semilunar attachments of the three leaflets of the aortic valve. The cross sectional diameter of the aorta at the nadir of the leaflet attachment where the aorta and ventricular muscle meet, and at the upper limit of the attachment at the sinutubular junction, are very similar, with the leaflets supported with a spatial relation as if to the sides of a cylinder. The diameter of the more distal circle at the sinutubular junction is, if anything, slightly smaller than the left ventricular outflow. This relation is lost in the Marfan syndrome. The aortic root becomes bulbous and the attachments of the leaflets are splayed out. The commissures are attached to an aorta of much greater circumference than at the nadir of leaflet attachment, the leaflets no longer co-apt, and the valve leaks centrally. The widest part of the aorta is in the sinuses of Valsalva where the echocardiographer picks up the very tips of the leaflets as the valve opens. The coronary orifices are displaced upwards as the aortic wall proximal to them dilates.
Now that we know that there are nearly as many molecular subsets of the disease as there are families, several earlier conundrums become clear. One is the occurrence of inherited aortic dilatation with a propensity for dissection in people who are skeletally unremarkable
The term form fruste was used for the situation in which the aortic root is characteristic of Marfan syndrome but other features were absent. With the new knowledge about the variation in the genetic abnormality from one family to another, it begins to make sense that there is clinical variability between families. There appear to be those at risk of dissecting an aorta with Marfan morphology but who look normal. On the contrary there are Marfan families with severely affected skeletons whose aortas are not particularly large and who do not dissect. A distinction must be made, however, between the very characteristic morphology of Marfan syndrome and the more funnel shaped aortic dilatation seen in hypertension and post-stenotic dilatation associated with bicuspid valves.
Framing disease
The concept of “framing disease” emphasises that our view of diagnostic categories changes with the scientific and clinical information that we use and may change with time, and is to a variable degree arbitrary. Thus in Marfan syndrome we could make the diagnosis purely by a particular bodily habitus or we could define the disease by an abnormality of fibrillin. The two diagnostic frames will not encompass identical patient groups.
A way of framing the disease9 that concentrated the attention on the aortic root while accepting that other features might or might not be present was to group Marfan cases within a diagnostic heading of annulo-aortic ectasia.10 It is probably a term best avoided. Anderson teaches us that the term “annulus” is incorrect in the case of the aortic valve because there is no ring (see accompanying article).11 There is another problem and that is that the measured diameter at the nadir of attachment of the aortic valve leaflets (the point loosely called the annulus) is characteristically normal in the presence of a very dilated Marfan aorta, even with severe regurgitation. It is the aortic sinuses that bear the brunt of the dilating process.
Should we make the morphological and histological abnormalities of the aorta our diagnostic criterion, since this is the focus of our surveillance and treatment, and not be distracted by a gallimaufry of clinical signs by which we decide whether the patient should or should not be included in the Marfan frame? Even here there is a problem because the degenerative change in the aortic media (loosely called cystic medial necrosis although not cystic and not necrotic) is the common end point of a number of aortic pathologies and is found both in hypertension and in the aortic wall beyond a bicuspid valve (fig1).
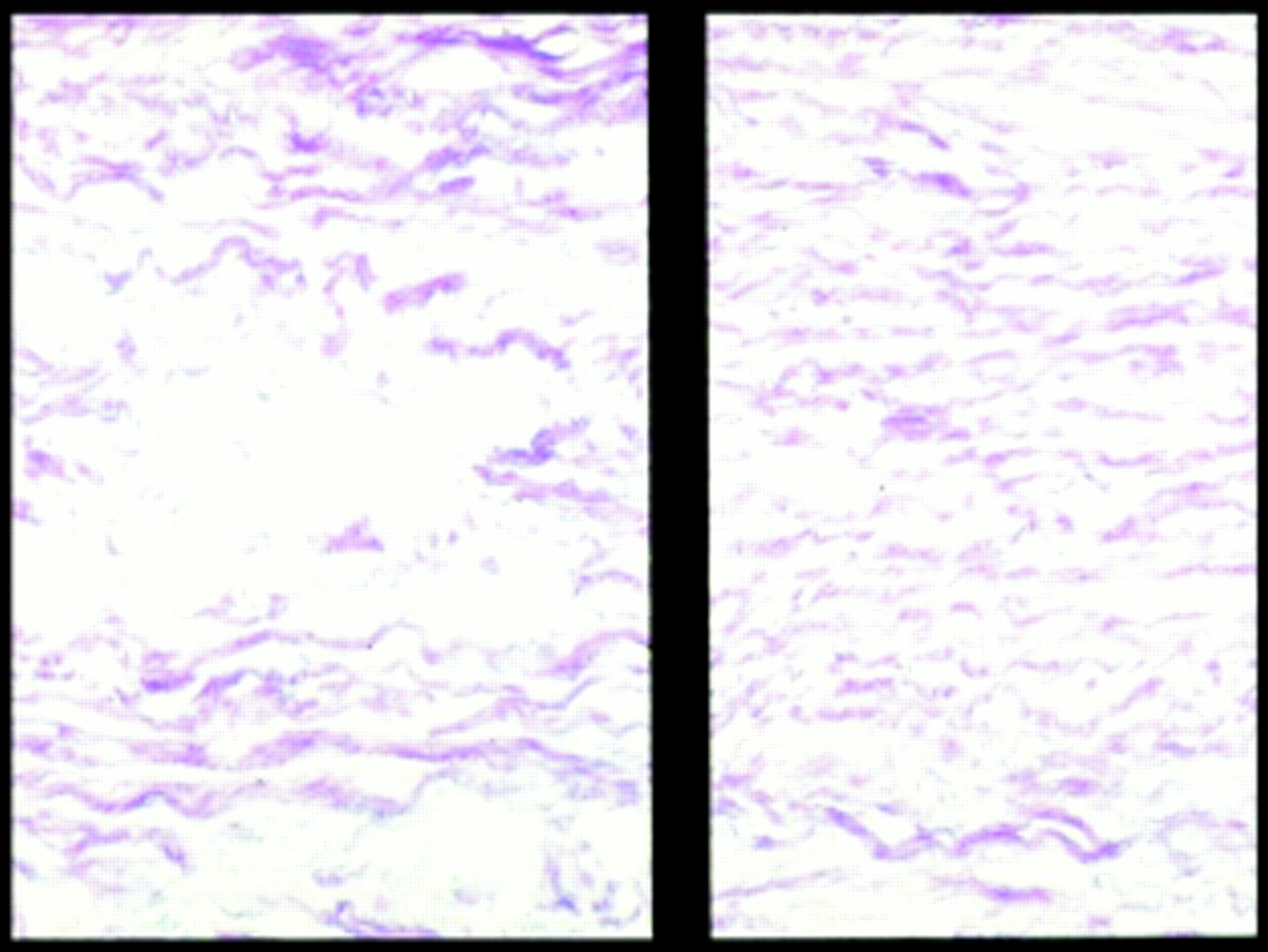
Histological appearance of a Marfan aorta. On the left is the severe form of medial degeneration called “cystic medial necrosis”.
Now that we know something of the gene involved in its inheritance, and of the structural and biochemical abnormalities that cause the collection of clinical features, need we carry on using the eponym, and indeed should we abandon the word syndrome?12“Dominantly inherited fibrillin abnormality” might be a better diagnostic frame.
The cardiovascular manifestations of Marfan syndrome can be viewed within any of these diagnostic frames, but in pragmatic terms cardiac surgical management is based on the morphology and size of the aortic root, the severity of aortic regurgitation, and its consequences for left ventricular function. The same is largely true of mitral valve disease associated with Marfan syndrome and with other abnormalities of the aorta. This diagnostic frame is the most appropriate in surgical decision making. This should come as no surprise. It is true of a number of other conditions that we treat. We treat aortic stenosis on its haemodynamic consequences almost independent of its aetiology, as we do most other examples of gross structural heart disease, congenital and acquired. Similarly, by the time a patient has end stage renal failure the management comprises renal replacement.9 The original causal factor is of diminishing importance.
Indications for operating on the aorta in Marfan syndrome
Emergency surgery
Dissection involving the ascending aorta is an absolute indication for operation to replace the aortic root in Marfan syndrome. It is said that at surgery there may be evidence of previous healed dissections, suggesting previous episodes of dissection have been survived. Indeed, there may be scars and stretch marks on the intima of the very attenuated sinuses of a Marfan aorta, but I do not believe that aortic dissection of the type we diagnose characteristically in these patients (figs 2 and 3) heals back to a subtle intimal lesion.

Classification of aortic dissection by the simpler dichotomous Stanford classification (ascending involved or not) and the older DeBakey system.

The typical form of lethal dissection, which is the most common cause of death in Marfan syndrome.
In Marfan syndrome replacement of the sinuses of Valsalva and as much of the ascending aorta as possible (or is practical) is the standard operation. In dissection caused by hypertension, or from unknown causes, the standard operation is repair of the dissection with a more conservative replacement of only as much of the aorta as seems necessary (fig 4A, B).

Operations for aortic dissection or ascending aortic replacement. (A) Simple tube replacement of the aorta for the sinotubular junction to the brachiocephalic origin. (B) Tube graft replacement and aortic valve replacement as separate components of the operation. (C) Composite graft replacement. (D) Leaflet sparing aortic root replacement.
Dissection of the descending aorta is managed as for any other aetiology with conservative hypotensive management and consideration of aortic replacement if there is ongoing expansion13 ,14 or leaking.
Elective root replacement
Elective replacement of the aortic root involves replacement of the sinuses, the entire aorta up to the innominate artery, and reimplantation of the coronaries. Bentall devised this operation in the 1960s when he incorporated a Starr-Edwards valve, hand sewn into a tube graft.15 Complete replacement of the ascending aorta was an impressive undertaking 30 years ago. Bentall's operation was used in its original form for some years with enface anastomosis of the coronary ostia to the graft and incorporation of the redundant aorta around the graft to contain bleeding from the multiple suture lines. While effective in containing what was a life threatening difficulty with the operation, this left the possibility of false aneurysm due to bleeding contained within the sack and a continued communication. The standard operation remains replacement of the root with a valved conduit but now most surgeons make neat button anastomoses of the coronary arteries, and resect the excess aorta and gain meticulous haemostasis under direct vision (fig 4C).
The most frequently used prosthesis incorporates a St Jude bileaflet pyrolytic carbon valve. These are factory produced, and the tube graft is impregnated with gelatin to prevent leakage.
There is growing popularity for valve conserving operations as promulgated by Yacoub and David (fig 4D).16 In this operation the aortic sinuses are resected down to a couple of millimetres above the attachment of the aortic leaflets. The tube graft is scalloped to match and the leaflets are functional within an aortic tube graft. The operation is based upon the belief that the leaflets themselves will not stretch and result in regurgitation. This may be true but the leaflets themselves are not free of histological abnormality.
The key question is one of timing of the operation in the life of the Marfan patient. Leaving aside the question of aortic valvar regurgitation, the purpose in operating is to pre-empt dissection. The ideal time to replace the ascending aorta would be one or two months before it dissects. The problem is that we cannot predict with any confidence when an aorta will dissect. There really are no premonitory symptoms or signs of any value. We rely on measurement.
Aortic root measurements are made according to strict protocol (fig 5). The measurement at the level of the tip of the valve leaflets, carefully made to reflect a true diameter, is the measurement on which we base our decisions.

Points of echo measurement for surveillance of the aortic root. The diameter of the aortic root can be measured at three different levels. In Marfan syndrome level B is critical.
We know that the larger the aorta the higher the risk. There is a general rule that an aneurysm larger than 6 cm has a greater than 10% chance of rupture within the next year irrespective of site and aetiology, and if that risk is higher than that of planned surgery, surgery is the safer course. In Marfan syndrome where experienced surgeons can perform operations at low risk, planned replacement of the ascending aorta was advocated at 5.5 cm and has now come down to 5 cm in some units. Advocates of early surgery must be mindful of two things. One is that zero mortality is an illusion in a small series. Nine in a row without a death (0%) suddenly becomes 10% if the next patient dies. The other is that operative mortality may be low but deaths in subsequent years from infection, false aneurysm formation, coronary anastomotic problems, anticoagulant related bleeding, and valve thrombosis have to go into the head count if we are to advise patients wisely. We continue to watch some patients indefinitely. Older patients with stable aortic dimensions may have declared themselves outside of the risk group for aortic dissection (fig 6).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
A patient who showed no increase in aorta size from the age of 44 to 54 years and an aortic dimension of around 40 mm. This patient does not reach any of our current criteria for root replacement.
Another factor in making the decision to perform elective root replacement is the rate of change. Progressive enlargement of the aorta from one clinic visit to the next is an ominous sign. It is difficult to make a rule but, as an example, if the aortic dimension changed from 4.3 cm to 4.6 cm then to 4.9 cm at six monthly visits, we would advise surgery based on the rate of change without waiting for the size to reach 5 or 5.5 cm.17
A family history of dissection influences the decision towards operation. Tendency for the aorta to dissect runs in families and the genetic evidence that there are differences between families and similarities within them lends extra weight to this view.
Finally we take into account aortic regurgitation. The criteria for valve replacement in aortic regurgitation are now well established.18 If there are symptoms attributable to regurgitation and/or evidence of an increase in left ventricular end systolic dimension, then valve replacement is indicated to protect the left ventricle and the patient's prognosis. These rules apply equally in Marfan patients. If surgery is indicated to correct aortic regurgitation it is likely that the aortic dimensions are at or near the accepted criteria and root replacement should be performed. In any case, if the patient merits valve replacement, it would be extremely unwise to simply replace the valve and leave a Marfan aorta. If the morphology is that of a Marfan aorta and there are no particular reasons to preclude it, we would replace the root under these circumstances.
Throughout I have placed emphasis on the importance of replacing the sinuses. However, there are occasions when there is aortic regurgitation and an enlarged ascending aorta. There may be doubt about the morphology of the aorta. If the dilation of the aorta is above the coronary orifices and is not of Marfan type, replacement of the aortic valve and ascending aorta separately (fig 4B) is an alternative. There is a surgical rule of thumb. If the distance between the attachment of the aortic valve leaflets (that is, the suture line for a prosthetic valve or valved conduit) and the coronary orifices is so small as to make composite root replacement difficult, it is unlike a Marfan aorta. Then the ascending aorta and the valve can be considered separately and the sinuses left with the coronary orifices. Characteristically the sinuses have grown so large in Marfan's that the coronary ostia are displaced upwards making re-implantation relatively easy, at least in so far as the length of the coronary arteries is concerned.
Descending aortic replacement
Elective replacement of the dilated but undissected descending aorta is uncommon but should be seriously considered if the dilated segment is 6 cm or if the aorta of the patient under surveillance is enlarging and reaches 5 or 5.5 cm. These are difficult decisions and should be made by a surgeon familiar with the problem and confident in operating in this area. If the aorta is generally enlarged and extensive surgery would be required for relatively small gain, it may be wiser to continue surveillance and hope to be able to deal with problems as they present. The chances of coming alive to urgent surgery are better in the descending aorta that in the ascending aorta. Conversely if there is a severe but localised dilatation or dissection, in a patient who is otherwise doing well, the benefits may greatly exceed the risks entailed.13 ,14
There are also those who have chronic descending dissection. These include the many who have been operated on for ascending dissection in whom residual dissection in the arch and descending aorta remains. There are also those who had an acute descending dissection (type B) who should remain under surveillance.
As more patients who have had a replacement for ascending aortic dilation become long term survivors, it is likely that we will see progressive dilatation in the descending aorta as years go by. The follow up of these patients should be in expert hands, either under an aortic surgeon or a cardiologist committed to the practice. A succession of different doctors staffing or training in the cardiological clinic cannot be expected to understand the issues.
We are now using endoluminal grafts (also know to cardiologists as “covered stents”) for a variety of aortic diseases. These will be considered in suitable Marfan cases.
Risk of paraplegia
In the best hands there is substantial risk of paraplegia at the time of surgery on the descending aorta. This ranges from a risk of under 5% for a localised resection of a saccular aneurysm to about 20% for extensive surgery to replace the thoraco-abdominal aorta.
The risks can be considered under two categories.
There is the anatomical risk. The extent to which the blood supply of the spinal cord is collateralised varies and is not predictable with any present techniques. In some there are very definite watershed zones. Paraplegia can result from the division of a single critical intercostal artery which gives rise to a key spinal artery. Of course, the longer the resected aorta the more likely that the blood supply of the cord will be anatomically compromised.
There is also a time related risk of infarction of the cord if its blood supply is interrupted by aortic cross clamping. This time related risk can be considered to be a physiological risk. To minimise this risk we use some form of bypass to the lower half of the body and keep periods of ischaemia as short as possible. The options for providing a blood supply to the lower half of the body during cross clamping are partial bypass (return to the femoral artery) or a heparinised (Gott) shunt from the arch to the femoral artery or, if the problem is localised, the descending thoracic aorta.
Mitral valve prolapse
Mitral valve prolapse should be assessed and treated as for any other cause of mitral regurgitation. If it is associated with aortic valve or root disease, the decision about timing and procedure is made more difficult. If there is mitral regurgitation meriting surgical correction and there is a margin of negotiation in its timing, it would be best if it could be done at the same time as the aortic surgery.
Valve conservation has the same advantages as for any other patient. However, these patients have many problems in their lives. My preference is towards mechanical valve replacement, particularly if surgery is being performed or has already been performed on the root and aortic valve. An opposite point of view would be biological solutions to spare anticoagulation throughout, in case the patient later needs multiple eye or spine operations.
The decisions about when to operate, what operative strategy to adopt, and the implication for anticoagulation are ones which I always share most fully with the patient and the family, including a written summary of the issues.19 Thinking time is essential.
There is reasonable evidence that β blockers slow the rate of dilatation and reduce the number of events.20 I suggest their use routinely. However, once the ascending aorta is replaced, if the remainder of the aorta appears stable the requirement for β blockers can be reconsidered. In the interest of the aorta, I advise against isometric forms of exercise.
Summary
Aortic dissection is the most common cause of early death in Marfan patients
Dissection can be averted by composite root replacement
Aortic dimension, rate of increase, and family history are the best predictors
Echocardiography is the best means of surveillance
Root replacement is a serious undertaking with continuing risk of complications