Article Text

Statistics from Altmetric.com
The field of coronary artery pathology is overwhelmingly dominated by one disease—atherosclerosis. It is therefore not surprising that insight into the onset of symptomatic coronary artery diseases mainly stems from patients with plaques. Coronary atherosclerosis may lead to relatively benign symptoms, such as stable angina, due to bare stenosis or restenosis of lesions. Life threatening symptoms (unstable angina, myocardial infarction or sudden cardiac death (SCD)) usually arise from plaque disruption and superimposed thrombosis, whereas additional arterial spasm or microembolisation of atherothrombotic materials may worsen the situation. But many non-atherosclerotic coronary diseases may become symptomatic due to similar flow limiting complications, which is clearly of importance for differential diagnosis (table 1).
Autopsy studies on large series of SCD victims—as have been carried out in the Veneto region of Italy, for example—have mapped the age dependent differences in coronary artery disease patterns. In children there is a predominance of congenital lesions and vasculitis (particularly Kawasaki disease),1 although atherosclerotic coronary death may also occur at young ages. Such examples of juvenile onset of atherosclerosis, albeit rare, illustrate the complex genetic backgrounds of the disease.2 In the adult population, myocardial ischaemia is only seldom caused by non-atherosclerotic pathology. Even in those cases, atherosclerosis is usually superimposed on (and sometimes masquerading as) the initial disease, since any significant injury or geometric change of coronary arteries (be it congenital, inflammatory or degenerative) accelerates development of atherosclerosis.
NON-ATHEROSCLEROTIC CORONARY ARTERY PATHOLOGY
Acquired pathology of coronary ostia
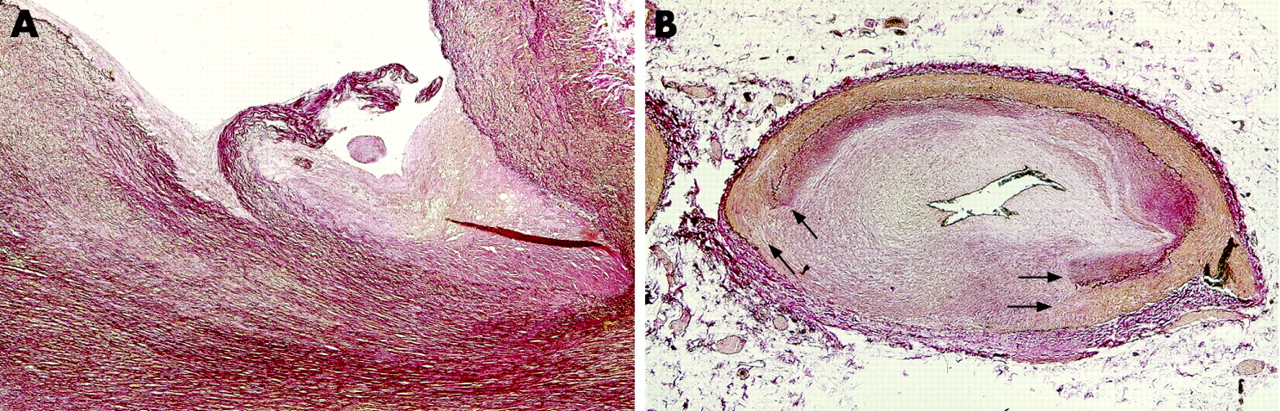
Every autopsy on the heart of a patient with suspected ischaemic heart disease should begin with inspection of the coronary ostia.3 Apart from atherosclerotic ostial narrowing, the origin of one or more coronary arteries can be compromised iatrogenically (in the case of aortic valve surgery or because of coronary catheterisation related trauma), or by coronary vasculitis or aortitis with extension to coronary ostia (Takyashu disease), by aortic valve endocarditis, by extension of aortic dissection, or by embolic obstruction (tumour, septic or atheroma). The flow limiting effects can be either acute (for example, in the case of trauma or spontaneous dissection), or otherwise have a later onset due to stenosing effects of a fibrocellular “wound healing” response to the injury (fig 1A).

Congenital anomalies of coronary arteries
A multicentre analysis of 7857 paediatric autopsy cases in UK, USA and Australia revealed an overall 0.5% incidence of congenital coronary lesions,4 indicating that although congenital coronary artery anomalies are rare, they can carry a significant risk of myocardial ischaemia. Anomalies relate to the origin, the course or the size of arteries, and occur either as isolated lesions or in the setting of more complex congenital heart disease. Some of these are relatively benign lesions, but others have an increased risk of sudden death in early childhood. The two most common isolated lesions considered as “high risk” in children are: (1) anomalous origin of a coronary artery from an abnormal sinus of Valsalva with an interarterial course between the great arteries; and (2) anomalous origin of one coronary artery from the pulmonary trunk. In the latter situation many patients die early, but those who survive develop exuberant collaterals between the artery arising from the pulmonary trunk and the artery supplied by the aorta. Aneurysmal dilatation and superimposed atherosclerosis compromises the patency of such vessels in the long term. However, it should be noted that minor coronary variations may also become significant later in life due to additional effects of atherosclerosis.5 This is illustrated by the so-called “high take off” coronary artery, which has its ostium several millimetres above the sinotubular junction. In this situation the artery may have a sharp downward angle and runs partially through the aortic wall; especially when there is additional atherosclerotic change of the ostium, such lesions may become symptomatic during effort.
Another interesting congenital lesion is myocardial bridging, indicating a segmental intramyocardial course of an epicardial artery. At autopsy myocardial bridging is a common finding, reported in 17–78% of human hearts, and as such should be considered an anatomic variant. Morales et al discriminated in victims of SCD between superficial and deep (2–3 mm) forms of tunnelled arteries, in which the latter was associated more frequently with regional fibrosis.6 Nerve-rich innervations of the myocardial bridge could contribute to profound systolic contraction that is characteristic for this lesion. Interestingly, myocardial bridging seems to protect against atherosclerosis, since the intramural arterial segment and the artery distal to a bridge are usually remarkably free of atherosclerotic plaque.
Coronary fistula
Arteriovenous fistulas are abnormal communications between an epicardial artery and a major vein (without interposition of the capillary bed). Fistulas between a coronary artery and pulmonary artery or cardiac chamber also occur, but are rare (<10% of all fistulas). Causes of fistulas are either congenital or acquired. In both instances, symptoms depend on the magnitude of the aberrant flow through arteriovenous communications, which can be single or multiple. Acquired fistulas have been observed as a complication of cardiac surgery, percutaneous coronary intervention (PCI) procedures or myocardial infarction. Congenital fistulas, similar to arteriovenous fistulas of soft tissue elsewhere in the body, can be clinically unrecognised at birth, but “grow up” with the patient and become symptomatic later in life. Eventually, angioma-like arteriovenous malformations composed of mature but malformed vessels develop, due to aberrant high flow in the coronary venous system. They should be distinguished from pure venous or capillary angiomas/vascular malformations.
Coronary artery dissection
Coronary dissection is not a pathological disease entity, but represents an acute complication initiated by pre-existent coronary artery disease or by traumatic coronary damage (or by a combination of both). Dissection of atherosclerotic arteries is very rare, but the incidence has increased drastically in the era of PCI and surgical coronary grafting. Up to 30% of all conventional balloon angioplasties result in angiographically visible coronary artery dissections, which are usually limited to the intimal plaque or the adjacent medial layer underneath the plaque. Such lacerations may heal through a fibrocellular intimal response. This tissue reaction (“wound healing”) is more excessive in cases of extensive laceration, and may increase the risk of subsequent restenosis.7 Perforation of the entire artery has been reported to occur in 0.3–0.6% of all patients undergoing PCI, but is not necessarily fatal. Cases of entire disruption of the vessel wall at the site of a previous PCI procedure may come up as an incidental finding at autopsy, in which the defect is filled up with fibrocellular tissue (“healed vessel rupture”) (fig 1B).
True dissection of the artery spreads longitudinally through the media, and requires at least some degree of pre-existent weakening of the medial layer. It is a serious complication, since as many as 80% of patients witness a sudden death. An example of connective tissue disorder causing weakness of the arterial media is Marfan disease, related to a mutation in the fibrillin-1 gene. The molecular defect causes cystic medial necrosis (CMD) of the arterial wall, characterised by fragmentation of elastic lamellae, apoptosis of medial smooth muscle and formation of proteoglycans-rich “mucoid pools”. In patients whose arteries are affected with CMD, a coronary dissection usually results from retrograde extension of an aortic dissecting haematoma. Patients with bicuspid aortic valve, which is basically a disease involving the entire aortic root, are also at risk from development of CMD of the aortic wall and the proximal part of the coronary arteries, and have a related risk of dissection. Other degenerative diseases which make the coronary artery prone to dissect, such as Ehlers–Danlos disease type IV and heritable storage diseases, are all extremely rare.
Most spontaneous coronary artery dissections occur in women of reproductive age (75% of cases), of which more than 30% are pregnant or peripartum.8 Progestogen-dependent connective tissue changes are held responsible for the association between dissection and pregnancy. There are several reports on infiltration of the dissected arterial segment with eosinophilic granulocytes; consequently, eosinophilic vasculitis has been proposed to explain the onset of dissection in these patients. However, it is arguable whether eosinophils should be interpreted as causative or as a reactive phenomenon (that is, an acute inflammatory response to the dissection).
Coronary vasculitis
Coronary vasculitis is rare but not short in variety, and may occur as an isolated form of vasculitis or as a manifestation of systemic diseases such as rheumatoid arthritis, systemic lupus erythematosus (SLE) or Behçet’s disease.9 Epicardial and small intramural vessels can be affected depending on the type of vasculitis involved. The most common form in children is Kawasaki disease, a self-limited type of vasculitis with acute onset and high affinity for coronary arteries, and one of the leading causes of acquired heart diseases in children. Skin, mucus membranes and lymph nodes of patients are also affected, but many patients with coronary artery involvement do not show this full spectrum of disease at the time of presentation. Infiltration of inflamed sites with immunoglobulin A secreting plasma cells, including coronary arteries, suggests an infectious trigger in the onset of Kawasaki disease.10 Around 15% of children develop coronary aneurysms which is, by far, the most important aspect of the disease. Aneurysms can cause rupture or myocardial infarction even in young children, but most lesions remain asymptomatic. Aneurysms may also develop in other forms of vasculitis, such as in polyarteritis nodosa (PAN) which affects coronary arteries in around 50% of patients, in giant cell arteritis which is usually a manifestation of a generalised arteritis at older age, and in rare cases of isolated coronary vasculitis. Persisting (post) inflammatory aneurysms are often multiple, and should be distinguished from true atherosclerotic aneurysms. Regression of aneurysms has been documented angiographically, but it should be noted that the phenomenon largely results from fibrocellular intimal proliferations which fill up the aneurysmal sac. With false (traumatic) aneurysms there is no dilatation of all three vessel layers like there is with true aneurysms; however, they show abrupt interruption of the vessel wall architecture by fibrocellular scar tissue. Such histological parameters are sometimes of importance for medico-legal reasons.
In many cases, histopathology of the vessel wall is not fully diagnostic for a specific type of vasculitis. For example, in children it may be difficult to distinguish Kawasaki disease from idiopathic juvenile onset PAN, since both diseases present with necrotising arteritis and aneurysm formation, and heal through stages of chronic inflammation and scarring. Another example is the presence of multinucleated giant cells in the inflamed coronary artery wall. Obviously, giant cells are a hallmark of giant cell arteritis, but the same type of cells can also be found in PAN, and in Wegener, rheumatoid or SLE vasculitis. In the advanced stages of atherosclerotic plaque development, giant cells are localised adjacent to the core of lipid debris amid foam cells. Therefore, final classification of vasculitis usually depends on combined interpretation of pathological, clinical and serological parameters. Lastly, coronary vasculitis should not be confused with an atherosclerotic inflammatory response.11 Distribution of the inflammatory cells throughout the arterial wall, composition of the inflammatory infiltrate, and association of mononuclear inflammatory cells with intimal lipid in the case of atherosclerosis, is usually helpful to differentiate between both diseases. Prominent adventitial inflammation also occurs both in vasculitis and in the advanced stages of atherosclerosis. Adventitial infiltrates are pathophysiologically of interest, because they contain well structured areas comprising B cells, dendritic cells and specialised endothelium, and as such closely resemble the mucosa associated lymphoid tissues (MALT) in the human body. These observations suggest that selection of B lymphocytes and production of (disease specific?) immunoglobulins takes place in the immediate proximity of the inflamed vessel wall.10 12
Non-atherosclerotic, non-inflammatory coronary artery diseases of particular interest
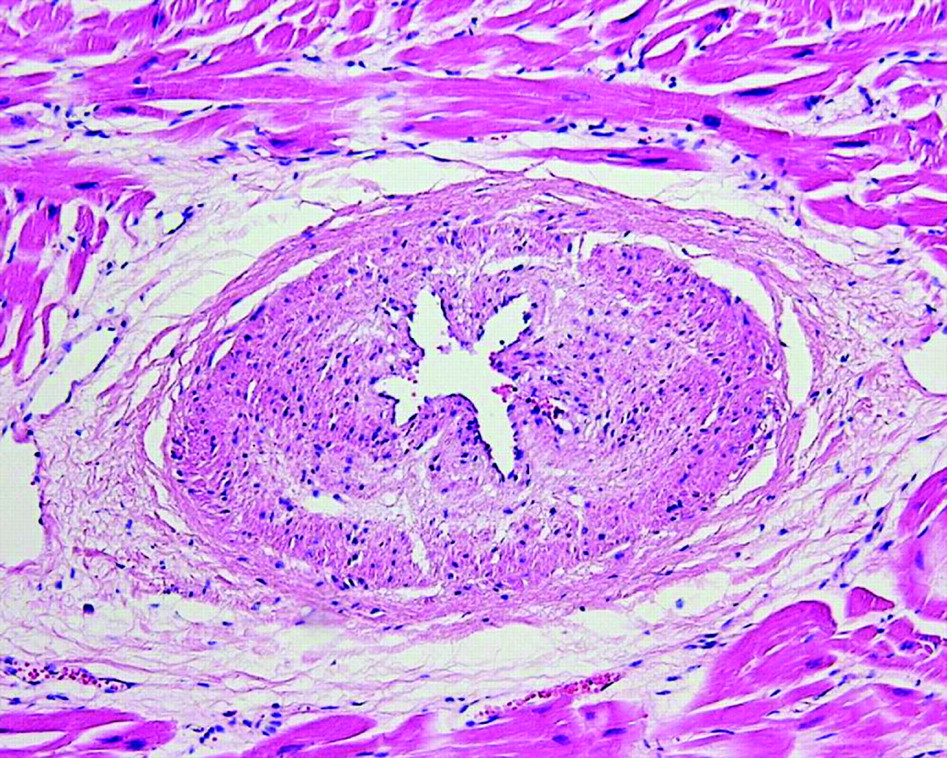
Fibromuscular dysplasia (FMD) is a segmental non-atherosclerotic, non-inflammatory vascular disease that commonly affects renal arteries. In the coronary artery bed the disease is rare, and if present, the small intramural arteries are involved more often than the epicardial arteries, although combinations may occur. Histologically, a subclassification is made into intimal, medial and adventitial types of FMD. The intimal type dominates in small intramural coronary vessels, and the fibrocellular proteoglycans-rich intimal cushions that characterise this disease lead to starshaped lumina on histology (fig 2). FMD of coronary arteries and arterioles has been described in association with many diseases including hypertrophic cardiomyopathy (HCM), Marfan syndrome, Friedrich ataxia, mitral valve prolapse and scleroderma.13 14 The strongest association is reported for HCM, in which 73% of affected hearts show large numbers of dysplastic vessels versus 8% of controls.15 The association of FMD with such a variety of diseases suggests a non-specific disorder of multifactorial origin. Nevertheless, FMC should be considered in the differential diagnosis of small vessel coronary artery disease (table 1), and particularly in cases of SCD in the young.

Mediastinal radiation therapy for malignancies causes significant coronary sclerosing intimal fibrosis in a minority of patients (up to 3% reported). This usually becomes manifest 5–10 years later. The intimal fibrosis is without an increase in intimal lipids. Bizarre shaped fibroblasts may accompany this fibrotic response, and there is usually adventitial fibrosis. These features may be helpful to distinguish from atherosclerosis.
Among the commonly abused drugs, cocaine and its derivatives (such as crack) specifically affect coronary arteries. Cocaine has spasmogenic and thrombogenic effects on the coronary artery wall, and in the presence of a fixed stenosis (atherosclerotic plaque), there is a serious risk of SCD.
CORONARY ATHEROSCLEROSIS AND THE VULNERABLE PLAQUE CONCEPT
Morphologic spectrum of “high risk” atherosclerotic plaques
Coronary plaque disruption followed by thrombotic occlusion is a widely accepted mechanism to explain the onset of most acute atherosclerotic coronary syndromes.11 Autopsy studies have revealed common anatomic features of ruptured plaques—a large lipid core covered by a thin cap which is heavily infiltrated with macrophages and T lymphocytes.16 These observations initiated the concept of vulnerable plaque: a subgroup of lesions with a high intrinsic propensity to develop thrombotic or haemorrhagic complications.17 However, coronary thrombus can be explained by a plaque rupture in only 60–70% of patients who present with acute myocardial infarction or sudden coronary death. Other thrombus initiating complications that have emerged from cardiac autopsies are plaque erosions, protruding calcified nodules and intraplaque haemorrhages.18 Furthermore, the composition of the underlying plaque differs in each situation, which implies that “vulnerable plaque” is not a single morphologic entity (table 2).
For example, advanced plaques with large conglomerates of dilated thin walled micro vessels predispose to intraplaque haemorrhages, whereas active inflammation in the fibrous cap of the lesion predisposes to plaque rupture or plaque erosion. Plaque rupture is undoubtedly the most powerful thrombus generating event. Many cases show protrusion of lipid mass with foam cells through the site of disruption into the lumen of the artery. This readily leads to massive activation of the coagulation system, largely due to very high tissue factor activity of foam cells. Indeed, up to 40% of thrombus masses aspirated from patients presenting with ST elevation myocardial infarction (STEMI) contain such cells.19 The thrombogenic stimulus in the case of plaque erosion is probably less dramatic and could underlie a mural thrombus in many patients with unstable angina or non-STEMI infarctions. But, in combination with pre- existing high grade stenosis, such erosions may also lead to acute coronary events including SCD.20 In a small but clinically well documented group of sudden coronary death victims <35 years of age (who had died of coronary thrombus), we found a plaque erosion underneath the thrombus in nine of 11 cases. This contrasts sharply with the much lower incidence of erosion as a cause of coronary death in the overall population.
Proper recognition of such diversity in the morphology of different vulnerable plaques and related onset of symptomatic coronary artery disease is clearly of importance, since most in vivo imaging techniques for detecting and treating vulnerable plaque are still focused on the rupture prone variant of vulnerable plaque.
Evolution of coronary thrombus
It is important to realise that plaque disruption and thrombus does not always imply a major symptomatic event, since many coronary thrombi remain mural and produce few if any symptoms. Significant plaque rupture leads to massive thrombotic occlusion by layered red thrombus, but smaller disruptions are initially covered with mural platelet thrombus (“white thrombus”). Such thrombi are fragile structures that either lyse spontaneously, or may wax and wane for some time, but bear the risk of distal embolisation (fig 3). After just 3–4 days the first steps of cellular thrombus organisation can be seen histologically, characterised by ingrowth of smooth muscle cells and overgrowth of endothelial cells, and later on by deposition of collagen and microvascular ingrowths. For a long time, only resident vascular wall cells were held responsible for organisation, but at present there is growing interest for an additional contribution of circulating mesenchymal stem cells in this process. Finally, the organised thrombus will be entirely incorporated into the plaque mass. Microscopically, such fully organised thrombi resulting from previous plaque rupture—so called “healed plaque ruptures”—have been identified in up to 10% of all plaques of patients with clinical coronary artery disease. Moreover, the number of old ruptures parallels an increase in lumen narrowing of plaques, indicating that clinically silent ruptures represent an important mechanism of (rapid) plaque growth.21

On the other hand, disrupted plaques can act as a stimulus for ongoing thrombosis, which ultimately progresses to occlusion. Histological staging of thrombus age in sudden coronary death victims (who died 6 h after the onset of symptoms) revealed fibrocellular ingrowth of several days old in their thrombus.21 Similar findings were documented by studying the histology of thrombosuction materials obtained from a large cohort of STEMI patients in the Amsterdam area; here, older (lytic or even organised) thrombus could be identified in 51% of 220 cases19 (fig 4). Such histological observations in living patients, which can be related strictly to the time of onset of acute coronary events, indicate an unpredictable time span between onset of plaque disruption and onset of clinical symptoms. Cellular and molecular mechanisms underlying variations in propagation and maturation of thrombus on top of such unstable plaques are presently not well understood.

Coronary microembolisation
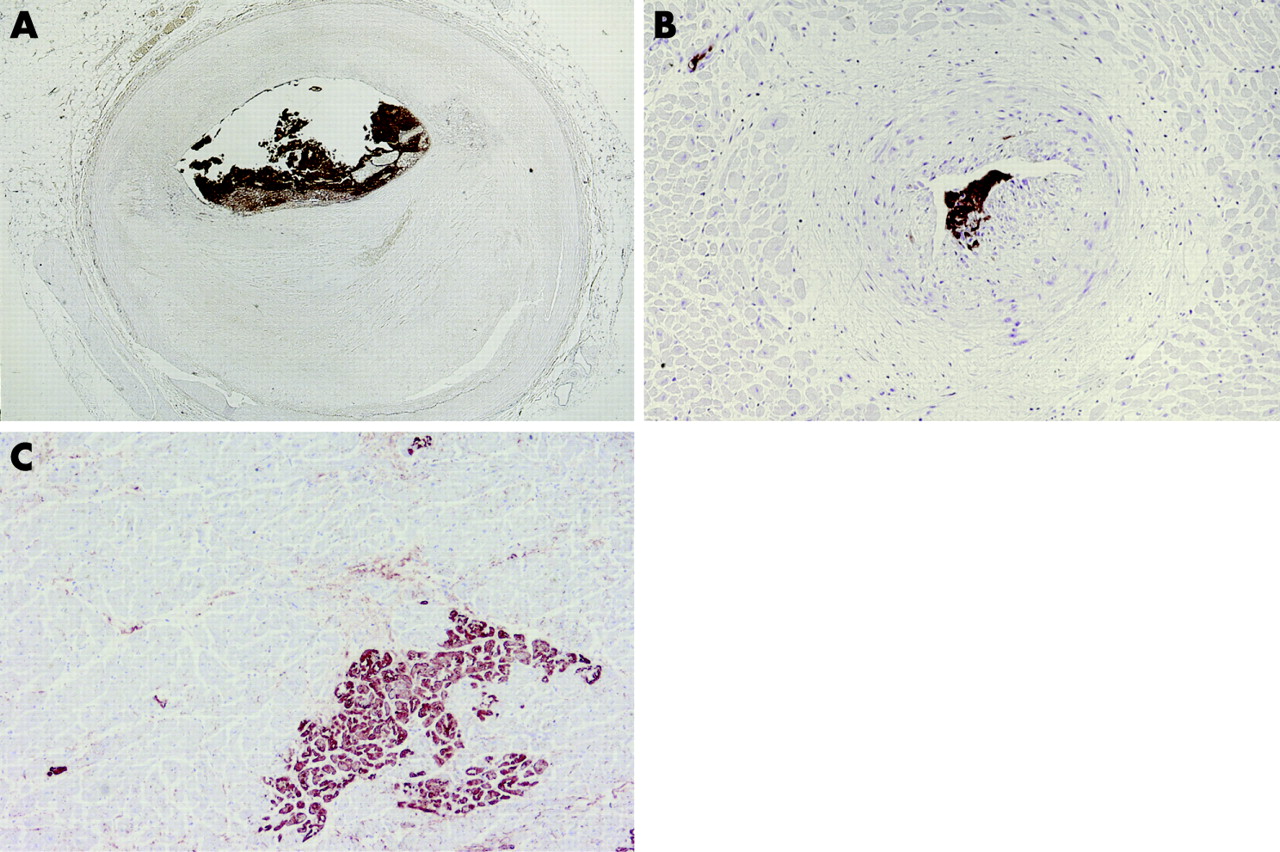
Every coronary artery disease complicated by thrombus may lead to spontaneous microembolisation of the distal arterial bed, especially when there is an aneurysmal component involved in the disease process. However, most cases of microemboli occur in the setting of atherothrombosis, which is further endorsed by presence of atheromatous gruel in many coronary emboli. Postmortem analyses of the myocardium of patients who died of ischaemic heart disease have revealed surprisingly high rates of microembolisation distal to thrombotic occlusions.22 Embolisation may complicate the process of spontaneous plaque disruption and thrombosis, but the risk increases during PCI when plaques are crushed mechanically. Important determinants for the occurrence and extent of microembolisation are the total atherothrombotic burden in the artery and the invasiveness of the interventional procedure. For example, stenting and coronary atherectomy cause more distal embolisation than balloon angioplasty due to severe plaque and/or vessel wall damage. Interventions in aorto-coronary saphenous vein grafts cause more emboli than in native arteries, due to larger plaque burden and the friability of (lipid-rich) plaques in grafts. Hence, high risk situations may benefit from the use of distal protection devices, which often contain large amounts of lipid debris (fig 5).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
CONCLUSIONS
Atherosclerosis can be diagnosed as the underlying disease in around 95% of symptomatic coronary disease patients >35 years of age, who have structural pathology in their arteries. It should not be forgotten that the remaining 5% harbour a huge variety of interesting iatrogenic, inflammatory, degenerative or congenital diseases, which can be potentially as dangerous as a severely stenosed or thrombosed plaque.
A major step forward in understanding coronary artery disease is the delineation and characterisation of a heterogenous subgroup of atherosclerotic plaques with high intrinsic risk for the development of acute coronary syndromes. This concept of “vulnerable plaques”, which is largely derived from autopsy studies, implies that the morphology of plaques is a more important denominator of clinical coronary disease than, for example, plaque volume or the degree of lumen stenosis. Clearly, this view has consequences for the development of new methods of in vivo coronary plaque imaging, but also for the treatment of patients with atherosclerotic coronary artery disease.
INTERACTIVE MULTIPLE CHOICE QUESTIONS
This Education in Heart article has an accompanying series of six EBAC accredited multiple choice questions (MCQs).
To access the questions, click on BMJ Learning: Take this module on BMJ Learning from the content box at the top right and bottom left of the online article. For more information please go to: http://heart.bmj.com/misc/education.dtl Please note: The MCQs are hosted on BMJ Learning—the best available learning website for medical professionals from the BMJ Group.
If prompted, subscribers must sign into Heart with their journal’s username and password. All users must also complete a one-time registration on BMJ Learning and subsequently log in (with a BMJ Learning username and password) on every visit.
REFERENCES
Footnotes
In compliance with EBAC/EACCME guidelines, all authors participating in Education in Heart have disclosed potential conflicts of interest that might cause a bias in the article. All authors declare that the answer to the questions on your competing interest form [http://bmj.com/cgi/content/full/317/7154/291/DC1] are all No and therefore have nothing to declare