Article Text

Abstract
Background COQ4 encodes a protein that organises the multienzyme complex for the synthesis of coenzyme Q10 (CoQ10). A 3.9 Mb deletion of chromosome 9q34.13 was identified in a 3-year-old boy with mental retardation, encephalomyopathy and dysmorphic features. Because the deletion encompassed COQ4, the patient was screened for CoQ10 deficiency.
Methods A complete molecular and biochemical characterisation of the patient's fibroblasts and of a yeast model were performed.
Results The study found reduced COQ4 expression (48% of controls), CoQ10 content and biosynthetic rate (44% and 43% of controls), and activities of respiratory chain complex II+III. Cells displayed a growth defect that was corrected by the addition of CoQ10 to the culture medium. Knockdown of COQ4 in HeLa cells also resulted in a reduction of CoQ10. Diploid yeast haploinsufficient for COQ4 displayed similar CoQ deficiency. Haploinsufficency of other genes involved in CoQ10 biosynthesis does not cause CoQ deficiency, underscoring the critical role of COQ4. Oral CoQ10 supplementation resulted in a significant improvement of neuromuscular symptoms, which reappeared after supplementation was temporarily discontinued.
Conclusion Mutations of COQ4 should be searched for in patients with CoQ10 deficiency and encephalomyopathy; patients with genomic rearrangements involving COQ4 should be screened for CoQ10 deficiency, as they could benefit from supplementation.
- Academic medicine
- molecular genetics
- muscle disease
- neuromuscular disease
- neurosciences
Statistics from Altmetric.com
Coenzyme Q (CoQ) is a vital molecule that transports electrons from mitochondrial respiratory chain (RC) complexes I and II to complex III. CoQ comprises a quinone group and a polyisoprene tail of varying length in different species, six in yeast (CoQ6) and 10 in humans (CoQ10). More than 10 nuclear genes (COQ genes) are involved in its biosynthetic pathway in yeast, for which there are homologous genes in virtually all eukaryotic species.1
In yeast, COQ gene products form a multienzyme complex organised around Coq4p,2 which does not appear to possess a specific enzymatic activity but plays rather a structural role. Human COQ4, on chromosome 9q34.13, encodes a ubiquitously expressed mitochondrial protein, which is able to complement the corresponding yeast deletion mutant functionally.3
Defects of COQ genes cause primary CoQ10 deficiency, a clinically and genetically heterogenous group of disorders, presenting as isolated nephrotic syndrome, as a multisystemic infantile disease, or as juvenile-onset ataxia, which are usually inherited as autosomal recessive traits.1 We now report a patient with CoQ10 deficiency associated with haploinsufficiency of COQ4.
Patients and methods
Case report
PT1 was born to healthy, non-consanguineous parents after an uneventful pregnancy. Weight, body length and head circumference were at the third percentile. He had dysmorphic features (epicanthal folds, broad nose, coarse facial features, syndactyly), a small ventricular septal defect, weakness, hypotonia and hyporeactivity. A standard karyotype performed at birth was normal. Tetrasomy 12p was suspected but was ruled out by karyotyping cultured skin fibroblasts. In subsequent months, growth was poor, hypotonia and weakness worsened and he had recurrent severe respiratory infections, which required hospitalisation. Serum lactic acid, ammonia and creatine kinase were normal. He did not have proteinuria and urinary organic acids were also normal. Because of the severe hypotonia a congenital myopathy was suspected and he underwent a muscle biopsy at another institution, which showed only increased subsarcolemmal succinate dehydrogenase staining in some fibres, and a prevalence of type I fibres, but no ragged-red fibres.
He was seen in our clinic at age 3 years. He had moderate mental retardation, was still unable to walk and did not speak. He was severely hypotonic and weak but had no other focal neurological deficit.
All studies have been performed with the informed consent of the parents. Skin fibroblasts had been obtained previously for the diagnosis of tetrasomy 12p syndrome.
PT2 and PT3 are the healthy parents of the previously reported child with CoQ10 deficiency caused by mutations in PDSS2.4
Methods
Array comparative genomic hybridisation analysis was performed using the Agilent 4×44K chip (Agilent, Santa Clara, CA, USA) according to the manufacturer's protocol. COQ4 sequencing, and real-time quantitative (RQ)–PCR analysis have been reported previously.3
Measurements of respiratory chain enzyme activities,5 CoQ10 levels,4 incorporation of radiolabelled para-hydroxybenzoate6 and fibroblast growth assays were performed as described.
Stable knockdown of COQ4 was performed in HeLa cells using specific small hairpin RNA expression vectors (HuSH pGFP-V-RS) purchased from OriGene (OriGene, Rockville, MD, USA) (shRNA sequences are available upon request). The vector contains a puromycin resistance gene and green fluorescent protein (GFP). After transfection cells were selected for puromycin resistance and by flow cytometry sorting GFP-positive cells.
COQ4 plasmid vectors and CoQ6 determination procedures have been described previously.3 6 7 Heterozygous strains for COQ4, COQ2 or COQ6 deletions were obtained by mating mat a haploid BY4741 deletion mutants, with mat α wild-type BY4742 cells. The mating and transformation procedures were performed as described.8 All biochemical analyses were performed after growth in non-fermentable medium containing glycerol (YPG).
Results
Genetic studies
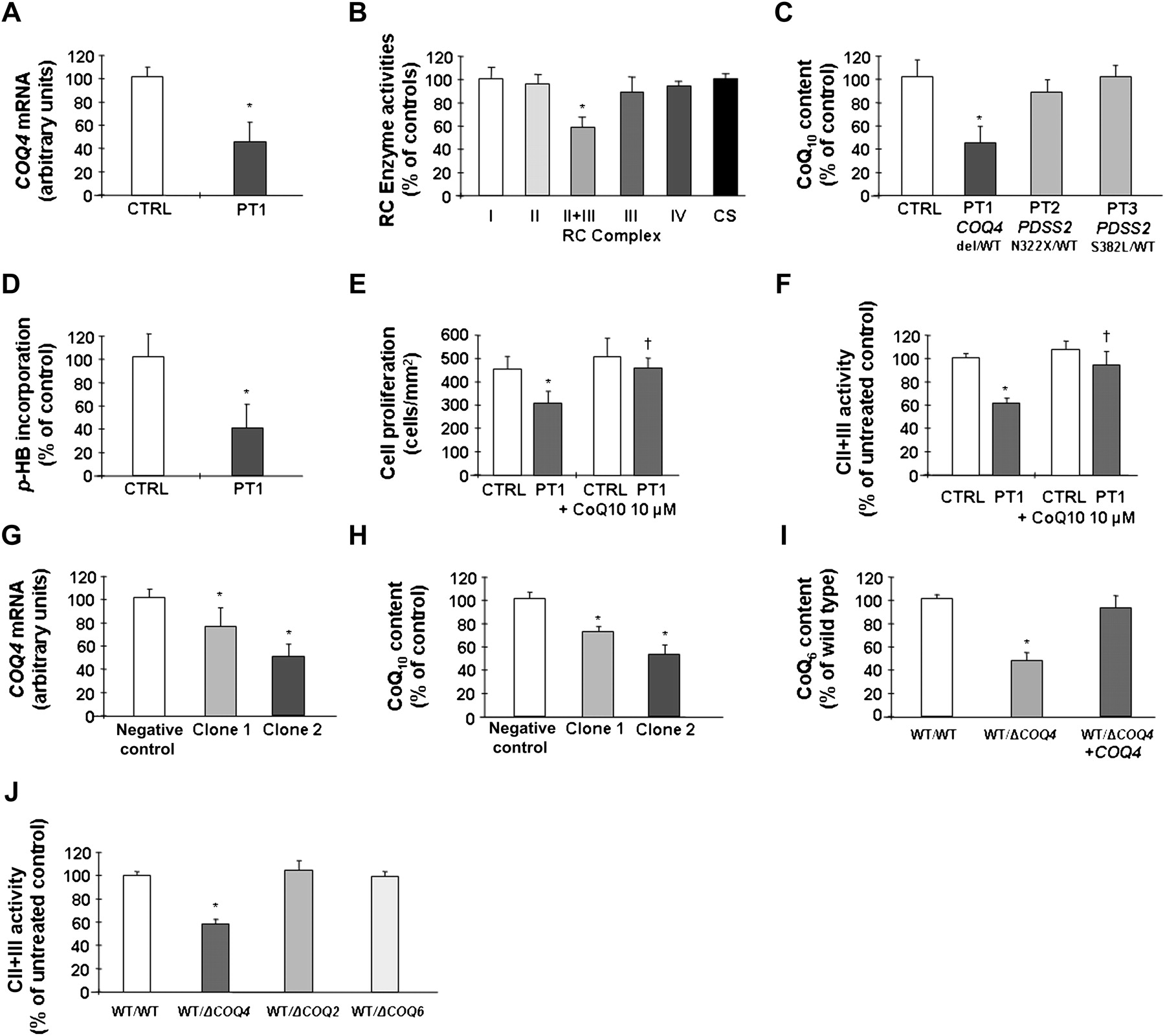
The presence of developmental delay and dysmorphic features was suggestive of a genomic alteration and array comparative genomic hybridisation detected a de-novo 3.9 Mb deletion of chromosome 9q34 (figure 1A,B) in DNA extracted from blood leucocytes. The deletion encompassed the COQ4 gene and its presence was confirmed in cultured skin fibroblasts by RQ–PCR analysis (figure 1C), but spares the STXBP1 gene. The supplementary table (available online only) lists all the genes included in the deleted region. COQ4 messenger RNA expression was reduced in these cells (48% of controls) (figure 2A), consistent with monoallelic expression. We ruled out the presence of a second mutation in the residual COQ4 allele by direct sequencing of both genomic DNA and complementary DNA.

(A) Array comparative genomic hybridisation (Agilent 44 K chip) of PT 1 showing the 3.9 Mb deletion: profile of chromosome 9 showing a series of spots having an about −1 log2 ratio at 9q34.11-q34.13. (B) Enlargement of the deleted region ranging from 129 531 to 133 523 Mb (assembly hg18). Oligos at 129 481 and 133 575 Mb resulted in normal log2 ratio. The quality of the experiment has been considered excellent on the bases of QC metric parameters (DNA analytics). The arrow indicates the position of the COQ4 gene within the deleted region. (C) COQ4 copy number in genomic DNA of cultured skin fibroblasts of the patient determined by real-time quantitative PCR.

{kind=link}
{kind=link}
(A) COQ4 mRNA expression and (B) respiratory chain enzyme activities, in PT1 cultured skin fibroblasts. (C) Coenzyme Q10 (CoQ10) content in PT1 cells and in cells of PT2 and PT3 who harbour heterozygous mutations in COQ1-PDSS2. (D) CoQ10 biosynthetic rate in PT1 cells measured by incorporation of 14C labelled p-HB. (E) Growth profile of PT1 skin fibroblasts. Incubation with 10 μM CoQ10 restores normal growth in patient cells. (F) Effect of CoQ10 supplementation on complex II+III activity in patient cells. (G) COQ4 mRNA levels and (H) CoQ10 levels in cells stably expressing an anti-COQ4 shRNA. (I) CoQ6 content in diploid yeast strains harbouring a heterozygous deletion of COQ4. Transformation with a plasmid-expressing COQ4 restores normal CoQ6 content in these cells. (J) Complex II+III activity in diploid yeast strains harbouring heterozygous deletions of COQ4, COQ2 and COQ6. Activities of other RC enzymes were normal (note that yeast does not have complex I). *Significant versus controls; †Significant versus untreated cells.
Biochemical analyses
Combined activity of complex II+III was reduced in fibroblasts (figure 2B), contrasting with normal activities of other enzymes, especially complex II and complex III and citrate synthase. These findings suggested CoQ10 deficiency. In fact, both steady-state levels of CoQ10 (43%) (figure 2C) and its biosynthetic rate measured by para-hydroxybenzoate incorporation (44%) (figure 2D) were decreased in the patient's fibroblasts.
Heterozygosity for PDSS2 mutations does not cause CoQ deficiency
We analysed CoQ10 levels in fibroblasts obtained from PT2 and PT3, the parents of a patient with PDSS2 mutations (N322X/S382L)4 who were both heterozygous carriers. In both cell lines, CoQ10 levels were normal (figure 2C), whereas the patient had CoQ10 levels less than 10% of controls.4
CoQ10 supplementation restores growth in patient's cells
Fibroblasts of PT1 displayed reduced growth compared with control cells, while supplementation with 10 μM CoQ10 restored a normal growth pattern (figure 2E) and complex II+III enzymatic activity in these cells (figure 2F).
Knockdown of COQ4 causes CoQ10 deficiency in HeLa cells
We generated HeLa cells stably transfected with a specific COQ4 shRNA vector. Analysis of two different clones showed a direct relationship between COQ4 mRNA expression levels (figure 2G) and CoQ10 content (figure 2H). In particular, clone 2 had approximately 50% residual COQ4 mRNA and 54% CoQ10, a situation that closely resembles that of the patient's cells.
Haploinsufficiency of COQ4 causes CoQ deficiency in yeast
Because COQ4 is highly conserved and the human gene effectively complements yeast COQ4null strains,3 we used Saccharomyces cerevisiae as a model to study the effects of COQ4 haploinsufficiency. Diploid yeast carrying a deletion of one COQ4 allele displayed a reduction of CoQ content relative to the wild-type strain, which was comparable to that observed in our patient's cells (figure 2I). Moreover, they displayed a similar reduction of the activity of complex II+III, 59±2% of the wild-type strain. Transformation with wild-type COQ4 rescued CoQ deficiency in these cells. We could not detect any defect in diploid yeast strains harbouring heterozygous deletions of COQ2 or COQ6 (figure 2J).
Effect of CoQ10 supplementation in the patient
After documenting CoQ10 deficiency in the patient, we started him on oral CoQ10 supplementation (30 mg/kg per day of ubiquinone). There was a clear improvement in muscle tone and strength, which became evident after 3 weeks of therapy. The patient began to walk unassisted after 5 weeks. The parents reported a general improvement not only in physical endurance, but also in his attention and social interaction. He also began to speak a few words. The improvement has been stable for 3 years; during this period he did not present with any severe respiratory infection. Interestingly, when the formulation was changed from phials to soft-gel capsules, the dosage was inadvertently reduced to 2 mg/kg per day of ubiquinone. After approximately 2 weeks the patient developed weakness and complained of diffuse myalgias. The dosage was immediately increased to 30 mg/kg per day of ubiquinone, with remission of symptoms within a week. Treatment was then switched to ubiquinol, the reduced form of CoQ, with a daily dosage of 15 mg/kg per day, without any significant problems.
Discussion
Primary CoQ10 deficiency is unique among mitochondrial disorders because early supplementation with CoQ10 can prevent the onset of neurological and renal manifestations.9 It is therefore of crucial importance to identify CoQ10-deficient patients and to characterise them at the molecular level.
While primary CoQ10 deficiency is usually inherited as an autosomal recessive disorder, our patient with haploinsufficiency of COQ4 also showed CoQ10 deficiency (both CoQ10 content and biosynthetic rate in cultured fibroblasts were reduced). The deficiency must have contributed to the clinical phenotype because CoQ10 supplementation resulted in a stable improvement of the clinical picture, and of the growth phenotype in cells. Moreover, when CoQ10 dosage was temporarily reduced to less than 1/10th of the effective dose, muscle symptoms reappeared.
In our patient, CoQ10 deficiency was milder than in other patients harbouring recessive mutations,10 consistent with the fact that one functional COQ4 allele was still present. Nevertheless, it has been shown that even partial CoQ deficiency in fibroblasts (in the range of 30–50% of controls) is accompanied by increased reactive oxygen species production and cell death.10
Knockdown of COQ4 in HeLa cells also resulted in a similar reduction of cellular CoQ10, with a direct relationship between COQ4 mRNA levels and cellular CoQ10 content. This situation appears to be unique for COQ4 because heterozygosity for mutations in other COQ genes such PDSS2 in humans (this report) and mice,11 or COQ7 in mice,12 does not cause CoQ deficiency. Taken together, these data provide further support to the notion that COQ4 has a critical function within the CoQ10 biosynthetic complex.
Haploinsufficiency of COQ4 in the wt/Δcoq4 diploid yeast strain was associated with a similar defect of CoQ6 content and of the enzymatic activity of complex II+III, which could be corrected by expression of wild-type COQ4. Heterozygous deletion of other COQ genes such as COQ2 or COQ6 had no biochemical effect, thus confirming that COQ4 is a limiting factor in the CoQ biosynthetic process also in yeast.
Deutschbauer et al,13 in a large-scale screening for genes displaying haploinsufficiency in yeast found that haploinsufficiency of COQ6 (but not of COQ4) resulted in a mild reduction of fitness. However, their results are not comparable with ours because they examined yeast growth in a medium containing glucose (YPD) that represses RC genes, while we performed our experiments in YPG, which forces the cells to rely on mitochondrial respiration and therefore stimulates the expression of RC genes. Another study showed that in yeast haploinsufficient genes are overrepresented in chromosome III,14 but COQ4 maps to S cerevisiae chromosome IV.
The clinical presentation in our patient was different from the phenotypes described in patients with mutations in other COQ genes (nephrotic syndrome, progressive multisystem disorder or juvenile-onset cerebellar ataxia) and more reminiscent of the two original patients with encephalomyopathy reported by Ogasahara et al15 in 1989. However, in our patient the phenotype was more complex as haploinsufficiency of COQ4 was associated with the deletion of at least 80 other contiguous genes in the region (see the supplementary table, available online only, for a list of the genes present in the deleted fragment). It is also possible that these associated defects might have increased the susceptibility of our patient to the effects of a relatively mild degree of CoQ10 deficiency. Other patients with CoQ deficiency and a predominantly myopathic phenotype include those reported by Sobreira et al,16 Di Giovanni et al,17 and Lalani et al,18 (who, however, still lack a precise genetic diagnosis) and the series of patients reported by Gempel et al,19 in whom CoQ deficiency was secondary to mutations in the electron transfer flavoprotein dehydrogenase gene. All these patients also benefited from CoQ10 supplementation, underscoring once again the importance of a prompt diagnosis of this condition.
To our knowledge, this is the first reported patient with CoQ deficiency treated with ubiquinol, the reduced form of CoQ that has become commercially available only recently, and that has a better bioavailability than the commonly used oxidised form (ubiquinone).20 Even though the dosage of CoQ10 is still administered empirically, the better bioavailability of the reduced form should allow physicians to employ lower doses of the compound (in fact 15 mg/kg per day of ubiquinol was sufficient to control symptoms), allowing a better compliance with treatment especially in children. Further research is clearly needed to compare the efficacy of ubiquinol versus ubiquinone in larger series of patients with CoQ deficiency, in order to optimise treatment protocols for this disease.
In conclusion, COQ4 should be considered a candidate gene in patients with CoQ10 deficiency and encephalomyopathy. Conversely, patients with genomic rearrangements of the 9q34 region involving COQ4 should be screened for CoQ10 deficiency, because supplementation could provide some benefit to their symptoms.
References
Footnotes
Funding This work was supported by Telethon Italy grant no GGP09207, CARIPARO foundation, the Spanish Ministerio de Sanidad (FIS) grant no PI 08/0500, University of Padova grant no 2010-CPDA102953, Italian Ministry of Health grant no GR-2009-1578914, National Institute of Health grant nos 1R01HD057543-01 and HD 32062, and Cariplo Foundation grant no 2007.5197.
Competing interests None.
Patient consent All analyses were performed with the informed consent of the parents of the patients. All analyses on patient fibroblasts were part of the standard set of investigations carried out to diagnose coenzyme Q deficiency.
Provenance and peer review Not commissioned; externally peer reviewed.