Article Text

Abstract
Objective This study aimed to investigate the efficacy of intrathecal morphine in the long term by hypothesising that a reduction of the intrathecal opioid dose following long-term administration would increase the level of pain intensity.
Design Randomised, double-blind, controlled, parallel group trial.
Setting Department of Pain Management, Russells Hall Hospital, Dudley, UK.
Participants 24 patients with non-cancer pain implanted with morphine reservoirs were assessed for eligibility.
Interventions Participants were randomly allocated to one of two parallel groups in which one of the groups had no change in morphine dose and the other group had a small reduction (20%) in dosage every week during a 10-week follow-up.
Outcome Primary outcomes were visual analogue scale (VAS) pain score change and withdrawal from the study due to lack of efficacy.
Results 9 of the patients assessed for eligibility declined to participate in the study. 15 patients were randomised to control (n=5) or intervention (n=10) and included in an intention-to-treat analysis. Owing to worsening of pain, seven patients withdrew from the study prematurely. None knew prior to withdrawal which arm of the study they were in, but all turned out to be in the dose-reduction arm. The calculation of dropout rates between groups indicated a significant statistical difference (p=0.026) and recruitment was ceased. The VAS change between baseline and the last observation was smaller in the control group (median, Mdn=11) than in the intervention group (Mdn=30.5), although not statistically significant, Z=−1.839, p=0.070; r=−0.47. Within groups, VAS was significantly lower at baseline (Mdn=49.5) than at the last observation (Mdn=77.5) for the reduction group, Z=−2.805, p=0.002; r=−0.627 but not for the control group (p=0.188).
Conclusions This double-blind randomised controlled trial of chronic intrathecal morphine administration suggests the effectiveness of this therapy for the management of chronic non-cancer pain. However, owing to the small number of patients completing the study (n=8), further studies are warranted.
Trial registration International Standard Randomised Controlled Trials Centre (ISRCTN 33733462).
- chronic pain
- drug delivery systems, implantable
- morphine
- randomised controlled trial
- treatment efficacy
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Article summary
Article focus
-
Recent systematic reviews were unable to find randomised controlled trials evaluating the effectiveness of long-term intrathecal drug delivery systems for the management of chronic non-cancer pain.
-
We aimed to investigate if a small decrease in the intrathecal morphine dose leads to an increase in reported pain scores in patients with chronic non-cancer pain undertaking long-term intrathecal morphine.
-
The randomised controlled trial design would allow investigation of the long-term efficacy of intrathecal morphine delivery.
Key messages
-
Statistically and clinically significant increases in pain intensity were observed for patients randomised to have intrathecal morphine dose reduction.
-
The findings of this study suggest the efficacy of intrathecal morphine delivery for the management of chronic non-cancer pain.
Strengths and limitations of this study
-
To our knowledge, this is the first randomised controlled trial investigating the efficacy of intrathecal drug delivery systems for the management of chronic non-cancer pain.
-
By investigating patients with intrathecal delivery for a minimum of 12 months, this study is not confounded by the need for dose titration and the non-specific psychological effects of a major intervention.
-
The limitations of this study include the small sample size as well as being conducted in a single centre.
Introduction
Opioid receptors were identified in the spinal cord in 1973.1 Subsequent animal studies demonstrated that intrathecal (IT) opioids produce powerful and highly selective analgesia.2 IT opioids exert their analgesic effect presynaptically and postsynaptically by reducing neurotransmitter release and by hyperpolarising the membranes of neurones in the dorsal horn, thus inhibiting pain transmission.3
The technique of intrathecal drug delivery (ITDD) is based on the principle that effective analgesia can be achieved by the action of some drugs at the dorsal horn and adequate concentrations cannot be achieved by systemic administration, or only by high systemic doses. Delivery of the drug by the IT route is a means of achieving these enhanced therapeutic effects. The smaller doses needed for IT administration also allow a reduction in side effects compared to systemic administration. Following the first clinical use of epidural4 and IT opioids,5 Cousins et al6 used the expression ‘selective spinal analgesia’ to describe the phenomenon that spinally administered opioids could produce a specific analgesic effect with few motor, sensory or autonomic side effects. It was subsequently demonstrated that the analgesic effect was, in the main, due to the uptake of the opioid directly into the spinal cord and cerebrospinal fluid.3
The key indications for ITDD systems are chronic pain unresponsive to curative medical or surgical measures and to more conservative palliative measures, including systemic analgesics, physical therapies, psychological therapies, perineural injection procedures and nerve lesioning procedure. Pathologies for pain are broad and only exclude psychogenic pains; they can be due to cancerous or non-malignant pathologies. Morphine is considered the ‘gold standard’ medication for ITDD systems because of its stability, receptor affinity and extensive experience of using the drug by this route.7
For chronic non-malignant pain, it is strongly recommended that patients have a comprehensive psychological assessment8 to: (1) assess possible concurrent psychopathology (eg, severe affective disorder, body dysmorphia and procedural fears) that might impede successful implantation and (2) consider what additional individualised preparation might be advisable for the patient.9 Cognitive behavioural therapy should not be excluded as a subsequent treatment option. It may ensure that the reduction in pain severity expected as a result of the ITDD system is capitalised on by the development of reduced pain related behaviours and increased activity in a range of adaptive behaviours.
The first reservoir for IT analgesic delivery was implanted in 1981,10 and since then continuous IT analgesia using opioids and other analgesics has become a recognised therapy for the management of severe and otherwise intractable chronic pain despite a lack of well-controlled studies. A 3-year prospective study of IT opioid treatment for chronic non-cancer pain showed that when patients with extremely severe pain problems are selected for ITDD, they are likely to improve with the therapy but their overall severity of pain and symptoms still remains high.11 At least minimally clinically important changes in pain intensity were observed in 95% of participants in a recent study with a mean follow-up duration of 13 years.12 Improvements were also observed in sensory and psychosocial outcomes.
Recent systematic reviews were unable to find randomised controlled trials (RCTs) evaluating the effectiveness of long-term ITDD systems for the management of chronic non-cancer pain.13 ,14 Overall, the use of IT opioid administration seems beneficial, but the current available literature is too sparse to draw definite conclusions mainly due to the quality of the evidence. A systematic review of multiple well-designed RCTs is considered the highest level of evidence for the efficacy of pain treatment, followed by a well-designed RCT of adequate size as the next best level of evidence.15 To our knowledge, there is only one such study of IT opioids and that is confined to cancer pain.16
In the absence of strong supporting evidence for the use of IT opioids for chronic non-cancer pain, the therapy must be balanced against its risks as procedure-related complications have been reported to occur at a rate of 0.29 events/patient-year and catheter-related complications at a rate of 0.05 events/patient-year.17 Possible infections include meningitis, epidural abscess, pump pocket infection or pump reservoir infection. The rate of meningitis reported by studies ranged from 2.3% to 15.4% and for wound infections from 4.2% to 8.8%.18 When considering only non-cancer pain studies, the percentage of patients with meningitis ranged from 0% to 4% and for those with wound infections, from 0% to 22%.19 Furthermore, less common but serious events of permanent neurological injury can occur due to the development of opioid-associated granulomata. The incidence for this adverse event has been reported as 0.04% after 1 year, increasing to 1.15% after 6 years.20 The management of different adverse events is varied as some acute side effects may resolve with time (eg, nausea, vomiting, dizziness or itching). Recommendations for aftercare, ongoing care, prevention and management of potential complications and side effects have been described previously.8 ,18
We had previously undertaken a prospective controlled study of single-dose morphine compared with saline in patients with chronic non-malignant pain and demonstrated spinal morphine to be efficacious in the short term for patients who responded to systemic morphine but in whom the side effects had become intolerable.21 The current study aimed to investigate the efficacy of IT morphine in the long term by hypothesising that a reduction of the IT opioid dose following long-term administration would increase the level of pain intensity. Our primary outcome was the visual analogue pain score change and withdrawal from the study due to the lack of efficacy.
Methods
Study design and participants
We conducted a single-centre, double-blind, equal randomisation (1:1), dose reduction, controlled, parallel group study. All participants provided written informed consent. The original protocol anticipated using diamorphine, but between the trial approval and start of the trial, the practice changed to using morphine and the protocol was amended to reflect this.
Treatment strategies for the management of chronic pain start with the lowest risk and least invasive intervention and progress if a treatment is not effective. The ITDD system is a last-resort treatment to treat severe chronic pain because of their invasive nature, concerns about long-term opioid use and the possible complications related to the procedure. It is considered for use in patients with chronic non-cancer pain after more conventional treatments have failed (eg, pharmacotherapy, transcutaneous electrical stimulation or, in some cases, spinal cord stimulation) and in those who respond to systemic opioids but where the side effects have become intolerable. Patient suitability is also determined by a multidisciplinary team assessment that includes a clinical psychologist. A biopsychosocial history is performed in which factors such as organic cause of pain, topography, duration of pain, pain intensity, coping strategies, social support, medico-legal matters, history of anxiety and/or depression, previous treatments, and drug and/or alcohol abuse are taken into consideration. Where there is a discrepancy across the clinical team comprising a physician, a physiotherapist, a psychologist and a specialist nurse, a case conference is set up to include the family physician and other psychologists, physiotherapists and physicians not directly involved in IT therapy.
Following multidisciplinary assessment, all patients have an inpatient trial of IT therapy prior to implantation. This is conducted by a repeated bolus of morphine and saline in a single-blind fashion.21 Patients reporting greater than 50% relief with morphine and less with saline are selected for the ITDD systems. Chronic dosing is extrapolated and titrated at refills (approximately two per month initially). A small increase in the opioid dose may be necessary to maintain adequate pain control. Recent observations indicate that significant differences cease following year 3 of therapy suggesting stability.12 Additional IT drugs were added if the level of analgesia is inadequate as per the polyanalgesic consensus conference algorithm.22 Adjuvant IT medication, such as bupivacaine, may contribute to achieving better pain control and maintaining low IT morphine doses in patients with cancer23 and without cancer.24
Eligible participants were adults aged 18 years or over with implanted IT reservoirs of programmable type (SynchroMed, Medtronic Ltd) receiving IT morphine for non-cancer pain and having had infusion for ≥12 months. Patients had reported a stable level of analgesia with the pump, based on their attendance for pump refills at which the dose did not change and they reported analgesia. In view of the need for weekly attendance during the study, only those patients living within a short time journey from the hospital, with access to transport and limited comorbidities, were considered.
The pain nurse approached eligible patients for consent and the patients were randomly assigned by computer-generated randomisation (PN) to one of the two parallel groups in which one of the groups had no change in the morphine dose (control group) and the other group had a small reduction (20%) in the preceding week's dose every week during participation in the study (intervention group). The allocation sequence was received in sequentially numbered, opaque and sealed envelopes to ensure that the sequence was concealed. Patients were unaware as to which group they were in, as the dose alteration or no change was conducted by telemetry with the screen not visible to the patient. The telemetry was conducted by a physician (JHR) who was the only investigator aware of the allocation. Pain scores and other outcome measures were collected by a researcher (RVD) blinded to the allocation of the patients.
Outcome measures
Primary outcome measures were the visual analogue scale (VAS)25 score for pain and withdrawal from the study. Secondary outcome measures were functional and psychological measures based on the Oswestry Disability Index (ODI),26 Hospital Anxiety and Depression (HAD) scale27 and Coping Strategies Questionnaire (CSQ).28 Participants were evaluated at baseline and each week during participation in the study. VAS and ODI were collected on a weekly basis. HAD and CSQ were collected fortnightly.
Patients were asked to rate their average pain intensity during the previous week using VAS. VAS consists of a 100 mm straight line with anchors at its ends labelled as no pain and worst pain imaginable. It is a recognised method for the assessment in variation of pain intensity.25 ,29 Clinically important changes were classified in accordance with a consensus statement that established a 10–20% decrease as minimally important, ≥30% as moderately important and ≥50% as a substantial change.30
ODI is used to assess the level of pain interference with various activities of daily living. It is a valid measure of condition-specific disability.31 ODI consists of 10 items/activities with six levels (range 0–5). The scoring of this questionnaire was calculated as recommended by Fairbank and Pynsent.31
The HAD scale is a self-report rating scale of 14 items with four levels (range 0–3). This scale is used to screen for anxiety and depression (7 intermingled items for each subscale). The total score for each subscale is the sum of the respective seven items (ranging from 0 to 21). The HAD scale is considered a valid instrument for detecting states of anxiety and depression.32
CSQ is a self-report instrument to assess the active and passive coping skills of patients with chronic pain.33 It includes cognitive coping strategies (diverting attention, reinterpreting pain sensation, catastrophising, ignoring pain sensations, praying or hoping and coping self-statements), behavioural coping strategies (increasing activity level) and effectiveness ratings (control over pain and ability to decrease pain). The scores of these subscales result in three factors that account for 68% of the variance in questionnaire responses (cognitive coping and suppression, helplessness, diverting attention and praying). This questionnaire is a valid and reliable tool for chronic pain patient assessment.28
Data analysis
An a priori power analysis based on a previous open study data of reduction in VAS for pain with IT therapy21 computed a sample size of 24 (12 per group) that would provide 80% power at the 5% significance level to detect a difference in the means of 1.2 SDs (unpaired t test) or a difference between the two proportions 20% and 80% (Fisher’s exact test). The power analysis was based on a study which compared one group receiving morphine with one group receiving placebo (saline). The difference in means in the pilot study (5.1–0.91=4.19) was not used as the basis for the power calculation as the difference in the pilot study was likely to be larger than the difference observed in the current study where both groups received morphine. A difference in the means of 1.2 SDs was considered as a realistic estimate since we allowed for the effect to be much smaller than that observed in the pilot study (2.6 SDs if the SDs of 1.3 and 1.9 are pooled). Imputation methods were not used since the dropout rate in the group randomised to have IT dose reduction was 70%. This high dropout percentage rate would bias the results regardless of the imputation technique employed. Therefore, we followed an intention-to-treat protocol; all participants were included in the analysis and this was limited to within-group and between-group comparisons of baseline and final observation scores.
The Kolmogorov-Smirnov test was performed to test normality of numerical data. The majority of the numerical data was not normally distributed and attempts to transform the data were unsuccessful. Therefore, differences between patient baseline characteristics were performed using the Mann-Whitney U test. Differences between the baseline and last observation scores were evaluated using the Wilcoxon signed-ranks test. Categorical variables were investigated using Fisher's exact test. Data were reported as the median (minimum–maximum). Statistical significance was judged at the 5% level. Statistical tests were performed using the Statistical Package for the Social Sciences (SPSS) software (V.19.0, SPSS Inc, Chicago, Illinois, USA).
Results
Between 2006 and 2011, 24 patients were assessed for eligibility and 9 declined to participate. Following inclusion in the study of 15 patients, it was observed that a high rate of patients withdrew from the research (figure 1). Owing to the large number of withdrawals, a first interim analysis was undertaken just beyond the halfway point, which revealed that the withdrawals were all from the group randomised to have dose reduction. The dropout rate in the group randomised to have IT dose reduction was 70% and there were no dropouts in the patients allocated to the control (no dose reduction) group. One participant left the study following week 1, three patients withdrew after week 2, two participants after week 5 and one patient after week 7. The IT opioid dose in the patients who withdrew from the study was reduced from a median of 1.6 mg/day (0.625–5.5) to 1.15 mg/day (0.4–2.8), which corresponds to a decrease of 36% (20–79) in the IT opioid dose. The reason for dropping out from the study was related to a worsening of pain for all the participants. Calculation of the dropout rates between groups indicated a significant statistical difference (p=0.026). Recruitment ceased at that moment.
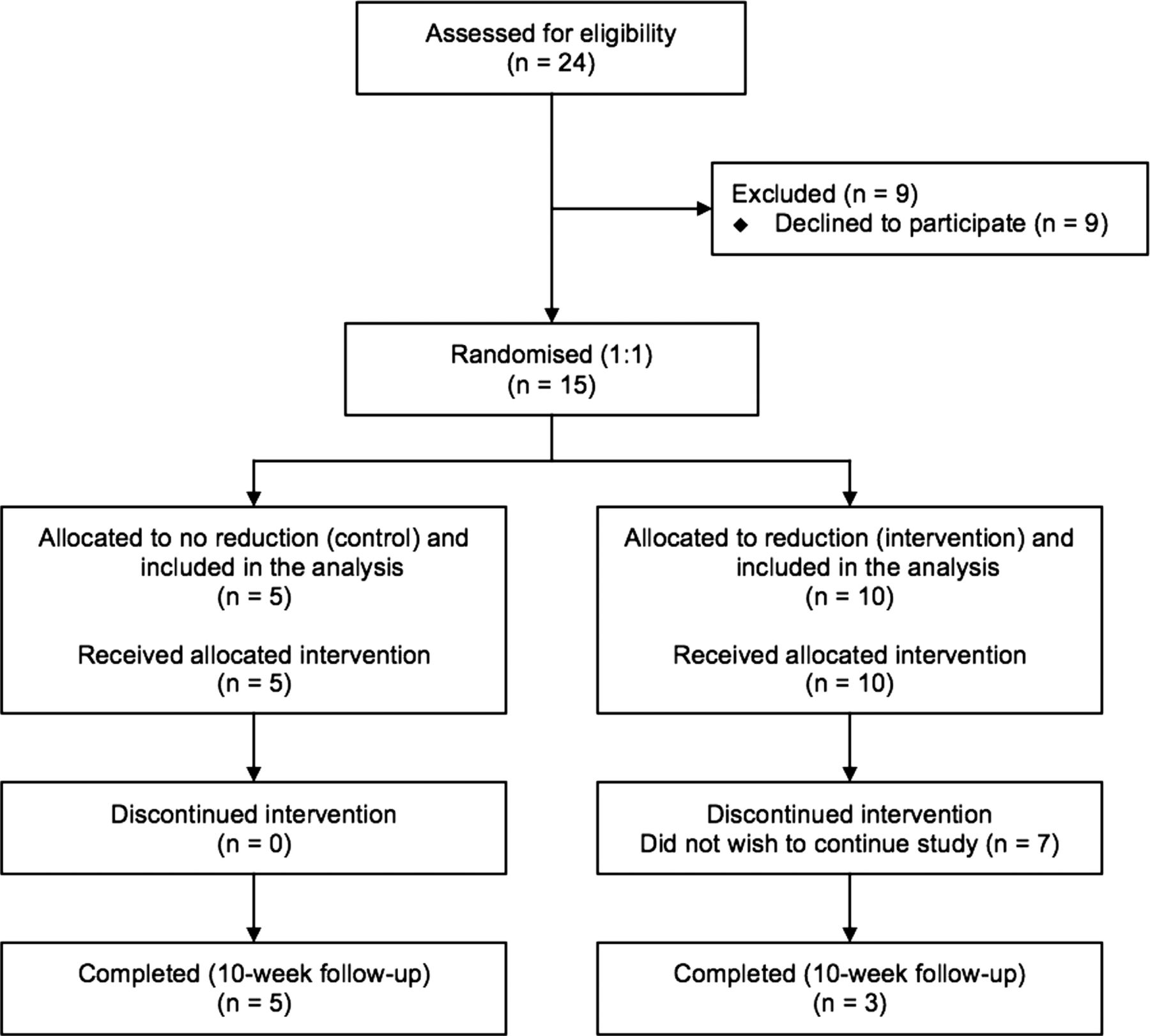
Flow chart of patient participation.
The patients recruited comprised eight men (53.3%) and seven women (46.7%) with a median age at the moment of enrolment in the study of 58 years (45–68). The median duration of the ITDD system therapy prior to participation in this study was 26 months (12–180). The pain syndrome was mechanical nociceptive caused by degenerative low back pain in five (33.3%) of the participants; visceral nociceptive due to postsurgery abdominal pain in one (6.7%) patient and mixed nociceptive–neuropathic following failed back surgery syndrome in nine (60%) participants. The five patients in the control group comprised two with mechanical back pain and three with failed back surgery syndrome; the 10 in the intervention group comprised 3 with mechanical back pain, 6 with failed back surgery syndrome and 1 with postsurgery abdominal pain. All patients had been on systemic opioids prior to pump implantation and thereafter only took opioids intrathecally. The preparations differed and the equivalent oral morphine dose prior to implant ranged from 20 to 240 mg morphine equivalent per day (tables 1 and 2).
Baseline characteristics of the patients according to randomisation groups
Baseline characteristics of the patients according to completion of study
There were no statistically significant differences between groups at baseline for age, gender, duration of therapy prior to study, adjuvant IT medications, VAS, ODI, HAD scale and CSQ (table 1). The IT opioid dose administered at study entry was significantly higher in the control group (median, Mdn=4.625) than in the intervention group (Mdn=1.612), a chance finding, U=7.00, p=0.028; r=−0.57. A comparison of baseline scores between patients who completed the study and those who did not demonstrates non-significant differences for all the variables, including the IT dose administered at the start of the investigation (table 2).
The VAS change between baseline and the last observation was lower in the control group (Mdn=11) than in the intervention group (Mdn=30.5), although not statistically significant, Z=−1.839, p=0.070; r=−0.47 (table 3). There were no statistically significant differences between the randomised groups in the changes detected for ODI, HAD scale anxiety and depression and all items of CSQ between the baseline score and the final observation.
Changes between baseline and the last observation
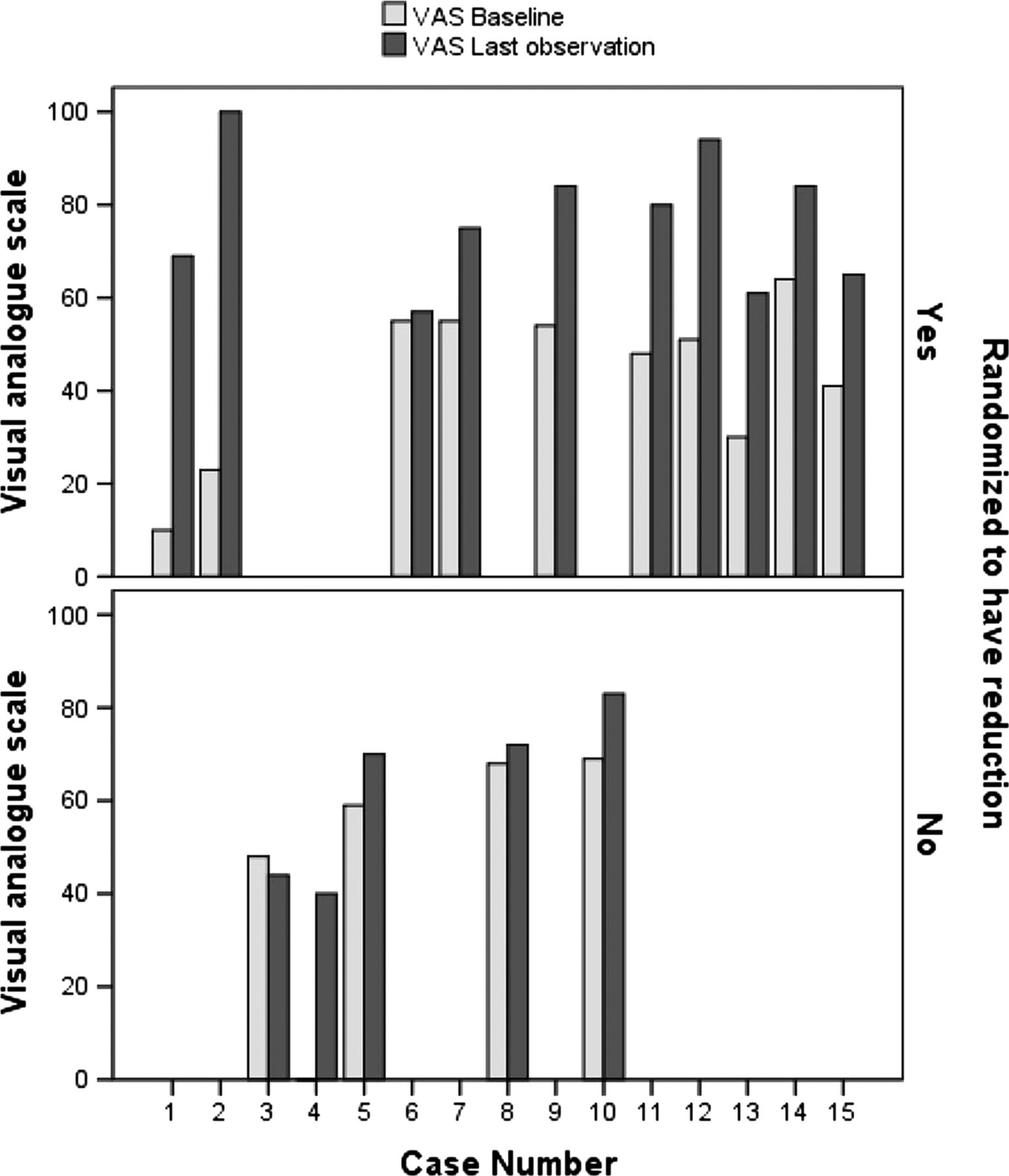
Within-group comparisons were also carried out (table 4). Statistically significant differences for VAS were observed between baseline and the last observation in the group randomised to have dose reduction (intervention) but not in the control group (p=0.188). VAS was significantly lower at baseline (Mdn=49.5) than at the last observation (Mdn=77.5) for the intervention group, Z=−2.805, p=0.002; r=−0.627 (figure 2). The ODI scores at baseline (Mdn=55.85) were significantly lower than at the last observation (Mdn=68.40) for the group allocated to have dose reduction, Z=−2.201, p=0.027; r=0.492. No statistically significant differences were observed for ODI in the control group (p=0.063). There were no statistically significant changes detected for HAD scale anxiety and depression and all items of CSQ in either randomised group between the baseline score and the final observation.
Within-group analysis for the visual analogue scale (VAS) and Oswestry Disability Index (ODI)

{kind=link}
{kind=link}
Individual visual analogue scale scores at baseline and the final observation for the control group (n=5) and reduction group (n=10).
The calculation of clinical changes based on the VAS scores indicated non-significant clinical changes in 10% of the patients in the dose-reduction group (intervention); minimally clinically important changes (≥10% and <30%) were observed in 20% of the participants randomised to this group, moderately important increase in pain (≥30% and <50%) in 40% of the participants and substantially important increase in pain (≥50%) in 30% of the patients. For the group where the morphine dose remained the same (control), non-significant changes were observed in 40% of the sample, minimally clinically important changes (≥10% and <30%) in 40% of the participants and one patient (20%) had a clinically substantial increase in pain.
Discussion
This RCT of IT opioid therapy in chronic non-malignant pain has demonstrated differences in pain relief between dose reduction and dose maintenance. It lends support to the efficacy of this therapy, which until now has not been subject to controlled trials.
A power analysis indicated that 24 patients would need to be included in the study to obtain a power of 0.8; however, owing to the high number of withdrawals, we undertook an interim analysis in which we found that the withdrawals were all in the dose-reduction arm. The attrition rate of 70% in the group randomised to have reduction also indicates that the treatment seems to be effective. Statistically significant differences between the arms were observed and the study was stopped. Although not statistically significant, the VAS change between baseline and the last observation was lower in the control group than in the reduction group. Within-group VAS and ODI differences were statistically significant between baseline and the last observation for the intervention arm with statistically significant greater pain and worsened disability in the dose-reduction arm. Clinically important changes indicating an increase in pain intensity were observed in 90% of the patients randomised to dose reduction (intervention). These changes were moderately important (≥30% and <50%) in 40% of the patients and substantially important (≥50%) in 30% of the participants.
Significant differences between groups at enrolment were observed for the morphine dose. The dose maintenance group (control) was found to have a significantly higher starting opioid dose. This mirrored the statistically insignificant trend towards the longer duration of IT therapy. It is possible that this group had greater levels of pain than the intervention group for the same dose of opioid and/or that with a longer duration of therapy, the dose had increased with time, as a small increase in opioid dose may be necessary to maintain an adequate pain control and recent observations from our unit indicate that significant differences cease following year 3 of therapy, suggesting stability.12 When dose escalation occurs, it is usually due to tolerance, progress of the disease34 or opioid-induced hyperalgesia.35
All participants had stable levels of opioid delivery, as evidenced by no change in the delivered dose at recent refills before investigation, and all reported analgesia with comparable pain scores (VAS). In using percentage dose reduction in this study, we anticipated overcoming a potential bias from this. Furthermore, no significant differences were observed at enrolment between those who completed the study and those who withdrew before completion, indicating that the initial opioid dose did not impact on the dropout rate. We had purposely chosen a small decrease in dose (20%) to prevent the patients suffering from any withdrawal symptoms and none occurred. This parallels the experience of Rauck et al36 in a study of opiate reduction within the context of investigating ziconotide. In this study, there was a 3-week weaning period prior to entering the trial, and thus the weekly reduction in IT opioids would therefore be approximate to 30%. The weaning process was successful in 92.9% of the patients; only 14 dropped out due to the inability to tolerate withdrawal, adverse events, non-compliance or patient’s request.
This study has recognised weaknesses of small sample size as well as being conducted in a single centre. The sample size was inferior to the 24 patients indicated by the a priori power analysis as the study was stopped when an interim analysis was conducted due to the large number of dropouts and revealed significant differences for withdrawals between groups. There was an imbalance in the number of patients in each group. The patients were randomised as a single block of 24, thus ensuring that in a sample of 24 there would be 12 in each group. Randomisation of smaller blocks would ensure that there were equal numbers in each group for smaller sample sizes as well (eg, if we had used a block size of 6, we would have had equal numbers in each group after 6, 12, 18 and 24 patients had been randomised). With our single block of 24, the chance of getting a split as uneven as 10 and 5 after 15 patients was about 9%. This RCT was conducted in a single centre. Selection for therapy followed the national guidelines8; however, their interpretation may vary in clinical practice even within the same country in the psychosocial domains of pain. Dose-titration strategies may differ across treatment centres. Different centres have reported average doses of 4.7 mg/day at an average of 3.4 years,37 7.42 mg/day at 29.14 months,38 9.6 mg/day at year 139 and 12.2 mg/day at year 3.40 This may lead to different levels of opioid delivery for which the sensitivity to dose reduction may differ.
The strengths of this study were not looking in the period following ITDD implantation because we considered that this period is confounded by the need for dose titration and the non-specific psychological effects of a major intervention. In investigating patients with IT delivery for a minimum of 12 months, we were able to focus on the evaluation of long-term efficacy of IT opioid therapy. To our knowledge, this is the first randomised, double-blind controlled study of this therapy in non-cancer pain. The findings of our RCT suggest the efficacy of IT morphine for the management of chronic non-cancer pain. Statistically and clinically significant increases in pain intensity were observed for patients randomised to have IT morphine dose reduction. In the light of these results, investigation of different populations and larger cohorts are recommended.
Acknowledgments
The authors would like to acknowledge the administrative and ward staff of the department of pain management.
References
Footnotes
-
Contributors JHR designed and was responsible for the conception of the trial. JHR, RVD, JLS, PN and GDK have made substantial contributions to (1) the acquisition of data, or analysis and interpretation of data; (2) drafting the article or revising it critically for important intellectual content; and (3) final approval of the version to be published.
-
Funding This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
-
Competing interests None.
-
Ethics approval The study was approved by the Birmingham and Black Country Research Ethics Committee (REC/35/02/JUN) and registered with the International Standard Randomised Controlled Trials Centre (ISRCTN 33733462).
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement No additional data are available.