Article Text

Abstract
AIMS To document survival rates after acute lymphoblastic leukaemia (ALL) during the era of modern chemotherapy, to assess effects of prognostic factors at presentation, and to investigate the relation of survival to patterns of organisation of care.
PATIENTS From a population based series of 5078 children diagnosed in the UK during 1980–94, 4988 remained for analysis after exclusion of nine children ascertained from death certificates alone and 81 who received no antileukaemia treatment.
MAIN OUTCOME MEASURES Actuarial survival rates.
RESULTS Between 1980–84 and 1990–94, the proportion of children treated at paediatric oncology centres rose from 77% to 89%, and the proportion entered into national trials rose from 59% to 82%. Each of age, sex, white blood count, immunophenotype, and Down’s syndrome status had a highly significant effect on survival. Five year survival improved from 67% in 1980–84 to 81% in 1990–94, a 42% reduction in the risk of death within five years of diagnosis. Survival did not differ significantly between hospitals with different numbers of new patients per year or between paediatric oncology centres and other hospitals. Children who were entered into national trials had higher survival and this difference became greater in recent years; five year survival rates for children diagnosed during 1980–84 were 70% and 64% for trial and non-trial patients, respectively; in 1990–94 the rates were 84% and 68% for trial and non-trial patients, respectively.
CONCLUSIONS Survival after ALL continues to improve. Nearly 50 children/year diagnosed during 1990–94 survived who would have died a decade before. Survival does not vary systematically with place of treatment but is higher for children entered into national trials.
-
Population based five year survival from acute lymphoblastic leukaemia has increased to over 80%
-
In Great Britain in the early 1990s, 50 children each year survived who would have died of the disease if diagnosed 10 years earlier
-
Survival of children with Down’s syndrome is comparable with that of other children
-
Survival did not vary systematically with hospital case load or between paediatric oncology centres and other hospitals
-
Children entered in national trials had higher survival rates than those not entered, and the gap has widened in recent years
- leukaemia
- survival
- referral patterns
- clinical trials
Statistics from Altmetric.com
The outlook for children with acute lymphoblastic leukaemia (ALL) has improved dramatically. In Great Britain, the five year survival rate increased from 4% among children diagnosed during 1962–66 to 75% during 1987–91.1 Patterns of organisation of care have also changed greatly. In the early 1970s, 20% of children were treated at hospitals seeing fewer than one new case of childhood ALL each year but 10 years later well under 10% were treated at hospitals with such a small caseload.2 In 1977, the UK Children’s Cancer Study Group (UKCCSG) was formed by a small number of consultants from seven hospitals. The group has expanded until it now covers 20 paediatric oncology centres in mainland Britain together with two in Ireland. The proportion of children in Great Britain with ALL who were initially referred to a UKCCSG centre rose from 64% in 1977–80 to 87% in 1989–92.3
A large part of the improvement in survival is clearly attributable to advances in treatment methods, particularly the increasing efficacy of combination chemotherapy. However, not all children benefitted equally from these developments. During 1971–84, higher survival rates were found among children who were treated at hospitals seeing larger numbers of cases of childhood ALL or who were entered in the trials.2 In the USA, a similar survival advantage was found for children treated at specialist centres or according to standard protocols.4
Most of the children in the 1971–84 study were treated before the start of the Medical Research Council’s eighth UK acute lymphoblastic leukaemia trial (UKALL VIII), which was open during 1980–84, and was the first childhood ALL trial in the UK to use relatively intensive chemotherapy.5 The effects on survival of treatment at centres with larger numbers of patients and of entry into trials were smaller in 1980–84 than in the 1970s, but in that study the period of follow up for the more recently diagnosed children was rather short.2 Since then, survivors who were treated in the early 1980s have been followed up for a further eight years and entry has been completed for another two large national randomised trials, UKALL X (1985–90)6 and UKALL XI (1990–96),7in each of which the best arm of the previous trial was used as the standard arm of the new one.
Our study was designed to document the population based survival rates for children in the UK with ALL during the era of modern chemotherapy, to assess the effects of prognostic factors at presentation readily available on a population basis, and to investigate the possible relation of survival to patterns of organisation of medical care.
Patients and methods
The National Registry of Childhood Tumours is the largest population based specialised childhood cancer registry in the world. It has been described in detail elsewhere.8 Our study is based on the 5078 children in the registry with ALL diagnosed during 1980–94. Of these, nine were ascertained from death certificates alone and have been excluded from all further analysis.
For each child in the study who received treatment for ALL, the treatment centre was defined as the hospital initially responsible for directing chemotherapy. Where care was shared between a tertiary referral centre and a local hospital, the tertiary centre was counted as the treatment hospital. Hospitals were classified in two ways. First, they were categorised according to their mean annual number of patients in the study series, grouped as in the 1971–84 study as fewer than one, at least one but fewer than six, and at least six patients each year.2 Second, they were divided into UKCCSG and non-UKCCSG hospitals; children who were treated at hospitals where consultants became members of the UKCCSG since 1980 were counted as UKCCSG patients only if they were diagnosed after their hospital became a UKCCSG centre. Children entered into Medical Research Council trials and studies of treatment for ALL were counted as trial patients, but those who were entered in acute myeloid leukaemia trials and whose diagnosis was subsequently revised to ALL were counted as non-trial. Children with T cell ALL in the early part of the study period and those with mature B cell ALL more recently were eligible for the UKCCSG non-Hodgkin’s lymphoma trials,9-11 and any who were entered have been counted as trial patients.
There are several sources of follow up information. Death certificates for all persons dying before age 20 with a neoplasm mentioned are matched against the registry. Surviving children are flagged in the NHS central registers, which then provide death certificates for patients who die of any cause, together with notifications of emigration resulting in loss to follow up. For our study, all flagged patients not known to have died or emigrated were assumed to be alive on 31 August 1998. Only three children had no follow up information from any source.
Survival rates were calculated by standard actuarial methods. Differences between the survival curves were tested by log rank tests and the χ2 test for linear trend. In addition to analysing overall survival from the date of diagnosis, we also analysed survival within three successive periods after diagnosis to assess the importance of prognostic factors at different phases of the patients’ care. These were: (1) the three months immediately after diagnosis; (2) between three months and two years, to assess survival during treatment, but excluding patients who died early from treatment related causes or who failed to enter remission; (3) beyond two years, to study subsequent survival of patients who received a full course of treatment.
Results
Table 1 shows the distribution by calendar period of diagnosis, age, sex, white blood count, immunophenotype, and Down’s syndrome status for all 5069 children in the study series. The age standardised annual incidence of 33.6/1 000 000 was typical for Western industrialised populations. There was a modest increase from 31.9/1 000 000 in 1980–84 to 35.7 in 1990–94. The age distribution, with a marked peak at age 1–4, was again typical of that found in Western populations, as was the small male preponderance (M:F = 1.3:1). White blood cell count was recorded for 97% of all children but immunophenotype for only 88%. Among children with known immunophenotype, common ALL (including pre-B) was most frequent (80%) and mature B cell leukaemia least frequent (1.8%). Table 1 also shows the distribution of treated and untreated children. A total of 81 children (1.6%) received no antileukaemia treatment. The highest proportions of untreated children were among subgroups generally regarded as having a poor prognosis, most notably infants aged under 1 year, children with Down’s syndrome, and those with B cell ALL. The proportion of children remaining untreated decreased significantly with calendar period of diagnosis (χ2 = 8.96 on 1 degree of freedom (df) for trend; p = 0.0028).
Presenting characteristics of children registered with acute lymphoblastic leukaemia, 1980–94
Table 2 shows the treatment centres for all treated children. Only 6% were treated at hospitals seeing less than one new case of childhood ALL each year and 81% were treated at a hospital seeing at least six new cases each year. No hospital had an average of four to five new cases each year; the largest hospital treated 45 new cases each year. Overall, 84% of children were treated at paediatric oncology centres, including all the hospitals with annual averages of six or more children; the largest number of patients at a single non-UKCCSG hospital was 41, an annual average of 2.7. Younger children were more likely to be treated at hospitals with large numbers of patients or at UKCCSG centres. There was also a tendency for children with high white blood counts to be treated at these hospitals, but treatment centre type did not vary with sex, immunophenotype, or Down’s syndrome status.
Treatment centres for all treated children with acute lymphoblastic leukaemia (ALL) 1980–94
Table 3 shows the numbers of children entered into national trials. The proportion entered increased sharply between 1980–84 and 1985–94. Infants aged under 1 year and children with Down’s syndrome were less likely to be entered. Children with common ALL appeared to be more likely to be entered than those with other subtypes, but this result should be interpreted with caution because immunophenotype was less often known for non-trial patients. Children treated at centres with larger numbers of patients or at UKCCSG centres were also more likely to be entered.
Proportions of children entered into national trials
Table 4 shows the five year survival rates for all treated children, classified by age, sex, white blood cell count, immunophenotype, and Down’s syndrome status. The best prognosis was at age 1–4 and survival declined with age thereafter. Infants aged under 1 year had a very low survival rate. Boys had a lower survival rate than girls. Children with a high white blood cell count, immunophenotype other than common ALL, or Down’s syndrome had lower survival rates. Because of the interdependence of all these characteristics, they were analysed simultaneously to assess the significance of their effect on survival. Table 4 shows the results. Each variable had a highly significant influence on survival when allowance was made for the other four. In the first three months after diagnosis the survival rate for boys was actually higher than for girls, although the difference was not significant. The commonly observed lower survival for boys only emerged beyond two years after diagnosis. The results relating to immunophenotype should be treated with some caution because of the rather high proportion of children for which this item of data was unknown—12% overall, 15% for infants, and 24% for children with Down’s syndrome. It is clear, however, that the poor prognosis associated with T cell or mature B cell disease did not persist beyond two years. Otherwise the effects of the prognostic factors were broadly similar in the three successive phases after diagnosis.
Survival of treated children categorised by age, sex, white blood cell count, immunophenotype and Down’s syndrome status
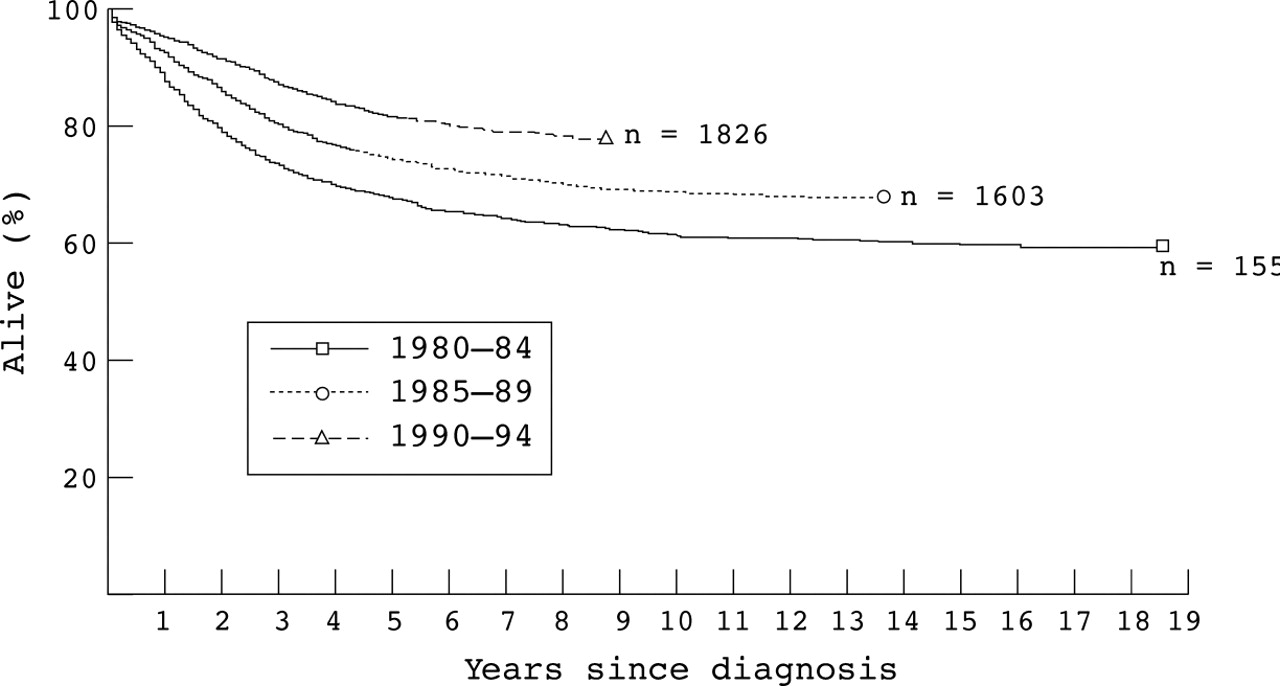
Five year survival rates for children diagnosed in successive calendar periods were 67% (SE, 1.2) in 1980–84, 74% (SE, 1.1) in 1985–89, and 81% (SE, 0.9) in 1990–94 (fig 1). The improvement in survival was highly significant overall (χ2 = 94.3 on 1 df for trend; p < 0.0001) and in each of the three phases after diagnosis; χ2 on 1 df for trend were 8.65 (p = 0.0032) for the first three months, 86.3 (p < 0.0001) for 3–24 months, and 17.2 (p < 0.0001) beyond 24 months.

Survival of children aged 0–14 years with acute lymphoblastic leukaemia first treated 1980–94, by calendar period of diagnosis.
Because the importance of prognostic factors can vary with treatment, we also analysed these separately for each quinquennium of diagnosis. Table 5 shows the changes in survival rate with period of diagnosis for children categorised by each factor. Overall, the effects of sex and white blood cell count were similar throughout the study period. However, the effect of sex on three month survival changed greatly with calendar period; survival was similar for boys and girls diagnosed during 1980–84 (95%) and 1990–94 (97%) but among those diagnosed during 1985–89 boys had a significantly higher survival rate than girls (97% v 95%; χ2 = 4.74 on 1 df allowing for other factors; p = 0.030). Survival of infants aged under 1 year did not change between 1980–84 and 1985–89; their survival rate in 1990–94 was higher, but this was based on a small number of cases and the difference was not significant. The survival of children with Down’s syndrome greatly improved in the most recent period to reach almost the same level as that of children without Down’s syndrome.
Five year actuarial percentage survival rates (SE) by calandar period of diagnosis, with results of χ2 test for trend in survival with calendar period
Table 6 shows survival rates for children treated at different types of hospital and for trial and non-trial patients diagnosed in three successive five year calendar periods. Survival did not vary significantly between hospitals with different numbers of patients or between UKCCSG and other hospitals during any part of the study period. When the group of exclusively UKCCSG hospitals with at least six patients each year was subdivided into those with annual averages of 6–10, 11–15, 16–25, and 26–45, there was still no evidence of any trend in survival with hospital size. Non-UKCCSG patients in the most recent period had a lower three month survival rate but this was based on only nine deaths.
Survival of treated children by treatment centre and entry into national trials
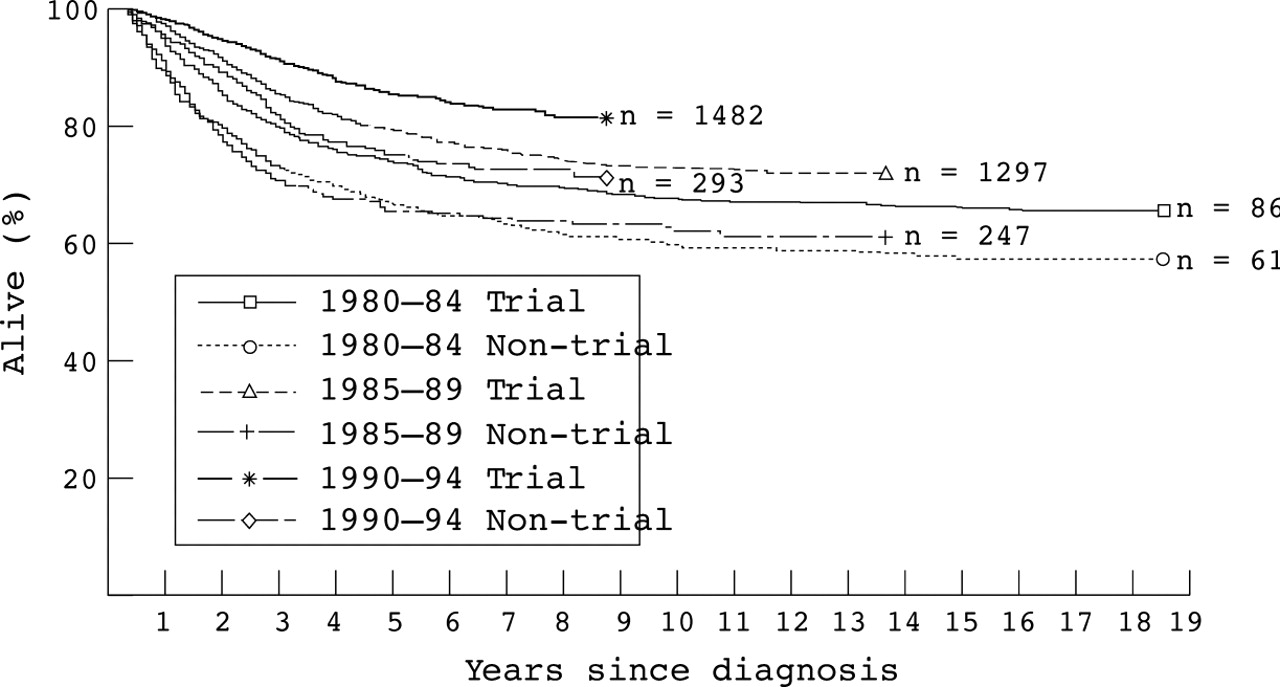
Overall survival was consistently higher for children who were entered into the trials. The difference was not significant for children diagnosed during 1980–84, but was significant for those diagnosed during 1985–89, especially 1990–94. In 1980–84, three month survival was significantly worse for trial patients, but early mortality was significantly higher for non-trial children in 1985–89 and 1990–94. Among children who survived at least three months, subsequent survival was consistently higher for those entered in trials during all three quinquennia (fig 2). Within the trials, there was no evidence for variation in survival with type of treatment centre during any part of the study period.
{kind=link}
{kind=link}
Survival of children aged 0–14 years with acute lymphoblastic leukaemia first treated in 1980–94, by calendar period of diagnosis and trial status. Deaths within three months of diagnosis have been excluded.
Discussion
By the early 1980s, the prognosis for children with ALL was already good but since then there has been further substantial improvement. Between 1980–84 and 1990–94, five year survival for all treated children increased from 67% to 81%, representing a reduction of 42% in the risk of death during the first five years after diagnosis; each year, nearly 50 children with ALL (∼ 15% of the total incidence) survived who would have died a decade before.
Previous analyses of prognostic factors in ALL have been based mostly on clinical trial data and none has included as many cases as our study. In common with other investigators, we found that children aged 1–9 at diagnosis and those with a white blood cell count of under 50 × 109/l had a better prognosis, and the combination of these two factors has been proposed as defining a good risk subgroup.12
Infants aged under 1 year at diagnosis had an especially poor survival rate. Our finding of no improvement during the 1980s corroborates on a population basis the similar finding of a study of infants treated on national protocols during 1980–90.13 In the most recent period, survival was higher, although not significantly so. The number of patients was small and this could be a chance finding, but it could also reflect greater success of the current protocol for infant ALL, which began in 1992.
Our finding that boys had a significantly lower survival rate than girls agrees with most previous studies.14 However, in the first three months after diagnosis boys had, if anything, slightly better survival than girls, suggesting that, while they have a higher chance of relapse, their remission rate is no worse than that for girls. The lower three month survival for girls diagnosed during 1985–89 is consistent with the significantly higher induction death rate for girls compared with boys in UKALL X.15 It was suggested that this might be a chance finding, and this is borne out by the return to similar short term survival for boys and girls diagnosed during 1990–94 in our study. As was found in UKALL VIII5and UKALL X,16 the survival advantage of girls only emerged about two years after diagnosis, indicating that boys are especially vulnerable to late relapse. It seems likely that this is related to the greater susceptibility of girls to myelosuppression during continuing chemotherapy.16
In analyses of Medical Research Council trial data, immunophenotype was not found to be an independent prognostic factor, the poorer prognosis for T cell and null ALL being explained by their tendency to be linked with other less favourable factors, especially white blood count and age.14 17 We found that, even after allowing for these factors, children with T cell, B cell, or null ALL had a worse survival rate than those with common ALL. The MRC data did not include mature B cell leukaemia, but in our series there was still significant heterogeneity of survival between immunophenotypes if B cell leukaemia was excluded, and this applied equally to trial and non-trial patients. A recent laboratory study suggests that sensitivity to specific chemotherapeutic agents varies between the principal immunophenotypes of childhood ALL.18
Children with Down’s syndrome and ALL have traditionally had a particularly poor prognosis. There was little sign of any improvement during the 1980s; the five year survival of ∼ 45% was higher than the 28% recorded in an earlier, but overlapping, series diagnosed during 1971–86,19 but a third of the children in that study received only minimal treatment. As more children with Down’s syndrome and ALL have received more intensive treatment their survival rate has increased dramatically and by 1990–94 was close to that for children without Down’s syndrome; however, the former still experienced a substantial excess of observed deaths over expected, largely because of the preponderance of other favourable prognostic features among children with Down’s syndrome. The most recent results still contrast greatly with those for acute non-lymphocytic leukaemia, however, where Down’s syndrome is now regarded as an indicator of low risk.20
During the 1970s, the survival rate for children with ALL was much higher if they were treated at centres seeing larger numbers of children with this disease. In our previous analysis of data for the early 1980s this effect was still present, although much diminished, but follow up was rather short and it has now been extended by eight years. In our present study, there was no evidence of variation in survival at any time from 1980 onwards with number of children treated. It seems likely that this is a result of treatment having become much more standardised at centres of all sizes.
In the 1971–84 study, children entered into national clinical trials also had a higher survival rate. Unlike the effect of place of treatment, the survival advantage associated with entry to trials appears to have become greater in recent years. This was partly because in 1980–84 the higher survival rate for trial patients beyond three months after diagnosis was counterbalanced by higher early mortality within the trials. Among children diagnosed during 1985–89, the excess mortality outside the trials was concentrated in the period three to 24 months after diagnosis, suggesting that entry into UKALL X reduced the risk of death during continuing chemotherapy. In 1990–94, in contrast, the advantage was greatest in the period immediately after diagnosis. The UKALL X protocol stipulated that patients should be notified at the start of treatment, but in UKALL XI notification was to take place within four to five weeks of diagnosis, and the recommendation that patients who died before the first randomisation (after the end of induction treatment) should be notified was dropped in the revised protocol of 1992. In our present study, three month survival for non-trial patients was significantly lower than for trial patients, both in 1990–91 (p = 0.0002) and in 1992–94 (p < 0.0001). Thus, it seems likely that in practice there was a bias against entry into UKALL XI of children who died early throughout the time that the trial was open. The difference in survival rates conditional on having survived three months seems much less likely to result from bias, and indicates that there was also a more long lasting advantage associated with trial entry during the most recent period.
A recent study found evidence of variation in survival between NHS regions of residence.21 Of the suggested explanations, differential access to centralised paediatric oncology services seems unlikely in view of our finding that survival was unrelated to centralised treatment, but the possible effect of regional variations in application of treatment protocols merits further investigation.
Acknowledgments
We are grateful to regional and national cancer registries, regional children’s tumour registries, the UK Children’s Cancer Study Group, the Clinical Trial Service Unit, the Office for National Statistics, the information and statistics division of the Scottish Health Service, and the Registrar General for Scotland for providing information on cases of childhood leukaemia and also to the NHS Central Registers in Southport and Edinburgh for notifications of deaths and for flagging survivors. We thank the many people who have worked on the National Registry of Childhood Tumours, particularly Ms AM Bayne, Mr MJ Loach, Mrs P Brownbill, Mrs J Williams, Miss A Berry, and Miss A Sabin. We thank Mrs EM Roberts for secretarial assistance. The childhood cancer research group is supported by the Department of Health and the Scottish Home and Health Department.