
Abstract
Preeclampsia and intrauterine growth restriction (IUGR) are two of the most common adverse pregnancy outcomes, but their underlying causes are mostly unknown. Although multiple studies have investigated gene expression changes in these disorders, few studies have examined epigenetic changes. Analysis of the DNA methylation pattern associated with such pregnancies provides an alternative approach to identifying cellular changes involved in these disorders. We analyzed methylation of 1505 CpG sites associated with 807 genes in 26 placentas from early-onset preeclampsia (EOPET), late-onset preeclampsia, IUGR and control subjects using an Illumina GoldenGate Methylation panel. Thirty-four loci were hypomethylated (false discovery rate <10% and methylation difference >10%) in the early-onset preeclamptic placentas while no and only five differentially methylated loci were found in late-onset preeclamptic and IUGR placentas, respectively. Hypomethylation of 4 loci in EOPET was further confirmed by bisulfite pyrosequencing of 26 independent placental samples. The promoter of TIMP3 was confirmed to be significantly hypomethylated in EOPET placentas (P=0.00001). Our results suggest that gene-specific hypomethylation may be a common phenomenon in EOPET placentas, and that TIMP3 could serve as a potential prenatal diagnostic marker for EOPET.
Similar content being viewed by others

Introduction
Preeclampsia is one of the leading causes of maternal and fetal morbidity and mortality, and affects 5% of all pregnancies.1, 2 It is characterized by high blood pressure in the mother and, frequently, growth deficiency in the fetus. Preeclampsia is heterogeneous in etiology and can be further subclassified into early-onset (<34 weeks) and late-onset (≥34 weeks).3 Intrauterine growth restriction (IUGR), even in the absence of preeclampsia, may show similar placental pathology (shallow invasion and remodelling of maternal vessels) and is also associated with significantly increased perinatal morbidity and mortality, as well as with disorders arising later in life.4, 5 Over decades, little progress has been made in the treatment and management of these disorders because they can only be diagnosed after full-blown manifestation of the condition is developed, by which time, treatment options are limited. Therefore, the identification of biomarkers that could be used to diagnose abnormal outcomes during early pregnancy would be a major step forward in antenatal care.
Although the exact cause is still unknown, epigenetic features are implicated in the pathogenesis of preeclampsia. Mutations in STOX1 were identified in some unique familial cases of preeclampsia with apparent maternal-only transmission of susceptibility.6 In addition, deficiency of the imprinted Cdkn1c gene in mice can lead to hypertension and proteinuria during pregnancy,7 further implicating the role of imprinted genes in the development of preeclampsia. Epigenetic alterations of non-imprinted genes have also been suggested to be involved. For example, the SERPINA3 promoter was found to be hypomethylated in preeclampsia-associated placenta,8 suggesting that the epigenetic alteration of this gene may be associated with reduced trophoblastic invasion and implicating this change as a potential biomarker for preeclampsia.
Many studies have investigated the gene expression profile in human placentas with preeclampsia and IUGR using genomic array technology.9, 10, 11 However, many factors may cause short-lived temporal changes in gene expression12, 13, 14 and, furthermore, placental RNA can degrade during parturition and rapidly after delivery of the placenta,15 making it difficult to obtain useful samples. DNA methylation is generally more stable and provides an alternative marker for underlying processes in the cell. In a previous study, we focused on the identification of highly variable ‘epipolymorphisms’ in the placenta. We then showed an association of one such epipolymorphism in TUSC3 with late-onset preeclampsia (LOPET), suggesting a role of altered DNA methylation in adverse pregnancy outcomes.16 In this study, we use the microarray data set to compare the patterns of DNA methylation in placental samples from pregnancies with and without preeclampsia and IUGR to search for potential biomarkers for these disorders.
Materials and methods
Sample collection
Fifty-seven placentas with or without associated preeclampsia and/or IUGR were collected from Vancouver BC Children's and Women's Hospital with informed consent from individuals, as was approved by the ethics committees of the University of British Columbia and the Children's and Women's Health Centre of British Columbia. Some data on these placentas have been previously published including analysis of trisomy in the placenta,17 analysis of altered imprinting for 11p15.5 imprinting control regions18 and an investigation of methylation variability in the placenta.16 Clinical information was collected on prenatal findings, pregnancy complications and birth parameters. Preeclampsia was defined as at least two of the following: (1) hypertension (systolic blood pressure ≥140 mm Hg and/or diastolic blood pressure ≥90 mm Hg, twice, >4 h apart) after 20 weeks, and proteinuria defined as ≥0.3g/day or ≥2+ dipstick proteinuria after 20 weeks, (2) non-hypertensive and non-proteinuric HELLP syndrome, using Sibai's criteria19 or (3) an isolated eclamptic seizure without preceding hypertension or proteinuria, using the British Eclampsia Survey Team criteria to define eclampsia.20 The preeclamptic placentas were subclassified into early-onset preeclampsia (EOPET) (onset before 34 weeks) and LOPET (onset at or after 34 weeks).3 IUGR was defined as either (1) birth weight less than third percentile for gender and gestational age using Canadian charts21 or (2) birth weight less than tenth percentile with either: (a) persistent uterine artery notching at 22+0 to 24+6 weeks gestation, (b) absent or reversed end diastolic velocity on umbilical artery Doppler and/or (c) oligohydramnios (amniotic fluid index <50 mm). All the LOPET and IUGR cases, 6 of the 17 EOPET cases, and 19 of the 32 controls overlap those used in our previous study of placental methylation variability.16 Detailed clinical information is provided in Supplementary Table 3. Although clinical details such as blood pressure and urine protein level were not always available in our controls, we excluded any cases reported as having hypertension or low birth weight.
Fragments of ∼1 cm3 were dissected from the fetal side of each placenta and DNA was extracted immediately after collection. Total RNA was extracted from five control placentas with two sites sampled from each placenta using RNeasy kit (Qiagen, Valencia, CA, USA) according to manufacturer’s instructions. Peripheral blood samples from normal individuals and fetal tissue biopsies (brain, kidney and lung) from abortuses were obtained with review board approval and were anonymous to individual identifiers.
Illumina microarray
DNA samples from 26 placentas were used for the DNA methylation array analysis. Samples were classified into three groups (EOPET, LOPET and IUGR) with their gestation-matched controls (Table 1). In addition, DNA samples from five additional control placentas with two sites dissected from each placenta were used to test for intra-individual DNA methylation variation. DNA samples extracted from blood of five normal female individuals and fetal tissues (brain, kidney and lung) from three abortuses were used to assess the tissue-specificity of methylation in the candidate loci. In all, 500 ng of genomic DNA was bisulfite modified using the EZ DNA Methylation Kit (Zymo Research, Orange, CA, USA) according to the manufacturer’s instructions. After bisulfite treatment, DNA samples were subjected to the Illumina GoldenGate Methylation Cancer Panel I array-based assay, which contains 1505 probes targeting 807 genes, using Illumina-supplied reagents and conditions.
Bisulfite converted DNA was mixed with allele-specific oligonucleotides in the assay, which target either the unmethylated cytosine (U) or methylated cytosine (C). A β-value of 0 to 1 was reported for each CpG site, which is related to the percentage of methylation, from 0 to 100%. β-values were calculated by subtracting background with the use of negative controls on the array and taking the ratio of the methylated signal intensity to the sum of both methylated and unmethylated signals. As a quality control step for Illumina array data analysis, we eliminated the probes with detection P-value >0.05 in any sample. To control for the possibility of methylation differences arising because of gender bias, we excluded all the probes on the X chromosome from our analysis. Differentially methylated loci between groups were identified based on the average DNA methylation level difference (Δ β) comparison and significance analysis of microarrays (SAM).22
DNA methylation and RNA expression of 10 placental sites from 5 normal term placentas were further assayed using the Illumina GoldenGate Methylation array and the Illumina Human Gene Expression array, respectively. Total RNA quality was verified and processed samples were hybridized to an eight-well microarray chip (HumanRef-8 v2). The BeadChip array was processed in the Centre for Molecular Medicine and Therapeutics BioAnalyzer Core Facility (Vancouver, BC, Canada). Output was analyzed using Illumina's BeadStudio software v3.2.7, 2007 (Illumina, San Diego, CA, USA).
Bisulfite pyrosequencing
Loci with absolute δ β >10% and false discovery rate (FDR) <10% in SAM were considered candidates of interest. To validate the differentially methylated loci identified from the Illumina array, bisulfite pyrosequencing was carried out for a subset of the candidate loci. In addition to the 26 samples run on the Illumina array, an independent set of 26 DNA samples from 13 EOPET and 13 control placentas were studied to validate the array findings. Pyrosequencing was performed on a Biotage Pyromark Q96 MD Pyrosequencer and the quantitative levels of methylation for each CpG dinucleotide were evaluated using Pyro Q-CpG software (Biotage, Uppsala, Sweden). All methylation-unbiased PCR and sequencing primers were designed to cover the same CpG sites interrogated by the Illumina probes (Supplementary Table 1). Methylation analysis of LINE1 elements was performed according to manufacturer's instructions (Biotage), as this measurement is commonly used as an indirect measure of global methylation.
Statistical analysis
Data from bisulfite pyrosequencing were analyzed with two-tailed Student’s t-test. Linear correlation was used to analyze the intra-individual methylation variation, the correlation between DNA methylation and gene expression, as well as the correlation between data obtained from Illumina array and bisulfite pyrosequencing assays.
Results
Unsupervised hierarchical clustering was performed on the Illumina GoldenGate methylation bead-array result from the 26 placental samples using the Illumina software and based on a distance measure of 1-r, where r is the Pearson correlation coefficient (Supplementary Figure 1). There was no obvious clustering of EOPET, LOPET, IUGR and control placentas. However, there was a preferential clustering of placentas according to gender (Supplementary Figure 2A), which is presumably caused by the inactivation of X chromosome in females (ie, higher methylation of X chromosome CpG islands in female than in male samples). After eliminating loci on the X chromosome from our analysis, the samples preferentially clustered according to their gestational age (ie, 83% samples with gestational week <34 clustered together and 76% samples with gestational week ≥34 clustered together) (Supplementary Figure 2B). In particular, only 2 out of 14 control placentas did not cluster based on this classification. These results suggested that gender and gestational age of samples were potential biases for DNA methylation analysis.
To eliminate these potential biases in the search of differentially methylated loci between placentas with and without adverse pregnancy outcomes, all Illumina probes on the X chromosome were excluded from our study. Furthermore, cases and controls were compared separately for each gestational age-matched group (ie, three comparison groups: EOPET, LOPET and IUGR, with their corresponding gestational age-matched controls). Within these matched groups, there was no significant difference of gestational age or maternal age (Table 1). Using a cut-off of <10% FDR from SAM, 192 loci were identified as being differentially methylated in EOPET as compared with controls, 16 loci in IUGR, but none in LOPET (Figure 1). As differences of small magnitude are less likely to be meaningful, we only considered differences between the mean methylation of patient and control groups of at least 10% absolute magnitude difference. Of the 192 loci with <10% FDR for EOPET, 34 had a methylation difference >10% (Δ β >0.1 from Illumina array) and all of them were hypomethylated in EOPET compared with the controls (Table 2). Of the 16 loci identified by SAM for IUGR, five had >10% methylation difference between controls and IUGR, all of them were highly variable in methylation value consistent with being a methylation allelic polymorphism – epipolymorphism as is commonly found in normal placentas.16

Venn diagram summary of differentially methylated loci. Differentially methylated loci were defined by a false discovery rate (FDR) <10% as calculated by significance analysis of microarrays (indicated as ‘SAM’) and average DNA methylation difference >10% as represented by ‘Delta Beta’. The number of differentially methylated loci is indicated in the overlapping area between circles. In all, 34 hypomethylated loci and 5 hypermethylated loci were identified in the EOPET group and IUGR group, respectively. *No differentially methylated locus was identified by SAM with FDR <10%. ↑: hypermethylated compared with the controls. ↓: hypomethylated compared with the controls.
To identify candidate sites at which methylation quantification could potentially be used for diagnostic purposes, it is important to select sites that are not greatly influenced by gestational age. Seven of the 34 candidate methylation changes associated with EOPET were significantly affected by gestational age as judged by comparing mean methylation in control placentas <34 gestational week as compared with controls ≥34 gestational weeks (AATK_P519_R, ABCB4_P51_F, CDKN1C_P6_R, EMR3_P39_R, FRZB_E186_R, PSCA_E359_F, SH3BP2_P771_R; Student’s t-test, P<0.05). From the remaining 27 loci, five sites (CAPG_E228_F, GLI2_E90_F, KRT13_P676_F, TIMP3_P690_R and MEST_E150_F) were selected for further validation by bisulfite pyrosequencing based on their magnitude of difference (GLI2, MEST and KRT13) and biological relevance to the preeclampsia development (CAPG, MEST and TIMP3).23, 24, 25
Bisulfite pyrosequencing validation of the five selected hypomethylated loci in EOPET showed that the Illumina array data correlate significantly with the pyrosequencing measurements for the same CpG site, as well as the mean of multiple sites in the pyrosequencing assays (Supplementary Table 2). To allow better representation of the methylation patterns in the associated regions, means of multiple CpG sites in pyrosequencing assays were used for all the comparisons. Therefore, an independent set of 26 placental samples, which consisted of 13 EOPET and 13 controls (gestational age was not significantly different between the two groups P=0.49; clinical information of the samples can be found in Supplementary Table 3), was analyzed by bisulfite pyrosequencing to confirm the differential methylation of the selected loci between EOPET and control placentas. All selected loci, except MEST (P=0.60) showed significant hypomethylation in EOPET (P=0.01 for CAPG, P=0.03 for GLI2, P=0.00003 for KRT13 and P=0.00001 for TIMP3) (Figure 2).

Box-plot of differentially methylated loci between EOPET and control subjects and their corresponding locations relative to the gene transcription start. Percentage of DNA methylation was assessed with bisulfite pyrosequencing for (a) CAPG, (b) GLI2, (c) KRT13 and (d) TIMP3 in 13 placentas with EOPET and 13 placentas without EOPET (indicated as ‘control’). P-values (indicated as ‘P’) were calculated by Student’s t-test. Locations for the differentially methylated regions targeted by pyrosequencing assays (regions bounded by vertical lines) are indicated in the box above the plots.
The most significant and largest absolute methylation difference (over 15%) was observed between EOPET and control at the TIMP3 locus. We tested the correlation in methylation values at this locus for two separately sampled sites from same placenta of five term control samples and these were well correlated with each other (R=0.90; P=0.038) (Supplementary Figure 3A). We further studied the methylation values at >10 sampled sites from each of two term placentas. The SD of methylation values in two placentas were only 2.5 and 1.6% (Supplementary Figure 3B), suggesting that there is little intra-placental variation of DNA methylation.
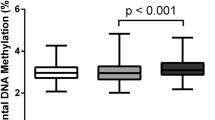
The feasibility of using placental DNA methylation for non-invasive prenatal diagnosis, requires differences to be present in comparison with maternal blood. From the Illumina array data, the TIMP3 locus was completely methylated in adult female blood and fetal tissues (99% methylated on average) with significant (P=0.001) differential methylation compared with the placental samples (73% methylated on average), which is comparable to the methylation differences observed for SERPINB5 (Figure 3), a marker that was previously suggested feasible for non-invasive prenatal diagnosis.26

Comparison of DNA methylation levels of TIMP3 and SERPINB5 between placentas, blood and fetal tissues. Both (a) TIMP3 and (b) SERPINB5 show lower methylation level in control placentas (five cases) than fetal brain (two cases), kidney (three cases), lung (three cases) and female blood (five cases). β-value of 0 to 1 represents the relative percentage of methylation from 0 to 100%.
Finally, we studied the relationship between the promoter DNA methylation and mRNA expression of the candidate genes in a subset of five term control placenta (two sampled sites each) that had been analyzed on the Illumina expression array. From the Illumina array data, we found that the DNA methylation of the TIMP3 locus was inversely correlated with its gene expression (R=−0.72; P=0.019), while none of the other three genes were significantly correlated based on this small sample size (N=10 samples) (Supplementary Figure 4).
Discussion
In spite of the many suggestions that epigenetic changes might be involved in adverse pregnancy outcomes,6, 8, 27 no genome-wide study has searched for epigenetic abnormalities in preeclampsia and IUGR. In this study, we profiled the DNA methylation of placentas from preeclampsia and IUGR pregnancies and their control counterparts using Illumina GoldenGate Methylation Cancer panel I array. Although the array mainly targets cancer-related genes, the pseudomalignant nature of the placenta makes it suitable for this study.28 Among the 1505 CpG loci targeted by the array, 34 loci were identified as hypomethylated in EOPET but none was differentially methylated in LOPET. The different epigenetic profiles in EOPET and LOPET placentas support the hypothesis that the two forms of preeclampsia are caused by different mechanisms.29, 30 EOPET, which is often associated with IUGR, is a severe form of preeclampsia (76% of our cases were associated with IUGR). It is suggested to be initiated by abnormal placentation, caused by reduced perfusion with increased apoptosis of trophoblasts.2, 30, 31 In contrast, LOPET, which is considered to be a maternal syndrome, is a mild form of preeclampsia. It is usually associated with normal placental development and a predisposed maternal constitution, such as hypertension or diabetes.2, 30
Epigenetic change may have a role in EOPET by altering gene expression and, as a consequence, normal placental development. Epigenetic changes may also result from hypoxic conditions associated with preeclampsia or an altered trophoblast composition in these placentas. Hypomethylation was found in many gene promoter regions in EOPET, but there was no difference in the global DNA methylation level as indirectly assessed by methylation at the LINE1 repeat sequence compared with other groups of placentas (data not shown). As LINE1 methylation is a measure of global methylation, these results imply that CpG hypomethylation observed is a gene-specific effect. Interestingly, many of the associated genes, such as the imprinted gene CDKN1C, are known to be important for normal placentation.32
To control for maternal and gestational age factors, the sample size used for array profiling in this study is small (8 to 10 samples per group). The small sample size likely explains why we do not find an association between polymorphic DNA methylation of TUSC3 and LOPET as we did in a previous investigation of over 100 placentas (P=0.02).16 This latter study was focused on the identification of epipolymorphisms and did not involve the statistical comparison of all methylation sites between groups, as was conducted for this study.
Recently, we reported a reduction of methylation at the H19/IGF2 imprinting control region in IUGR-associated placentas, but we did not find altered methylation at CDKN1C or other imprinted genes in IUGR and/or preeclampsia.18 The discrepancy can be attributed to the different ways of grouping samples, because we divided preeclampsia cases into PET and PET+IUGR previously without considering the effect of gestational changes on DNA methylation. Global changes of gene expression have been previously reported in association with gestational age.33 Our current findings suggest this is important also in regard to DNA methylation. Thus, the gestational age-dependent profile is important to evaluate and control for when considering any methylation change identified as a potential biomarker. This is particularly important in the study of preeclampsia as such pregnancies tend to be delivered early and comparisons with term births may be inappropriate.
The DNA methylation differences of CpGs in CAPG, GLI2, KRT13 and TIMP3 were confirmed in an independent set of 26 placentas with EOPET and gestational age-matched control pregnancies. Among these four genes, TIMP3 had the largest difference in DNA methylation level with an over 15% reduction in EOPET compared with control placentas. A previous study showed that TIMP3 gene expression can be regulated by promoter DNA methylation in the placental tissues.34 Although our assays target CpG sites upstream of the CpG island where previous groups analyzed (Figure 2d), we also found a significant inverse correlation between its DNA methylation and gene expression in placentas. Therefore, hypomethylation of the TIMP3 promoter may alter its gene expression in EOPET.
TIMP3 is a family member of the matrix metalloproteinase inhibitors, which have an important function in regulating a wide range of physiological processes such as cell growth, invasion, migration transformation and apoptosis. This gene is highly expressed in placenta and suggested to be important for implantation and decidualization by regulating trophoblast invasion.35, 36 Elevated expression of many TIMPs, including TIMP3, has been reported in preeclamptic placentas.37, 38 The hypomethylation of the TIMP3 promoter found in this study may increase TIMP3 expression and, in turn, reduce the invasiveness of trophoblast during placental development, which leads to placental hypoperfusion in EOPET. Intriguingly, hypermethylation of the TIMP3 promoter has been reported in choriocarcinoma and hydatidiform mole, conditions that have increased trophoblast invasiveness,34, 39 which further supports the inverse relationship between TIMP3 promoter methylation and trophoblast invasiveness. It has also been shown that TIMP3 could inhibit angiogenesis by blocking the vascular endothelial growth factor from binding its receptor,25 a well-known defect that is found in the trophoblast of preeclamptic pregnancies.40 Although the cause of the epigenetic modification is unknown, it may be related to the hypoxic environment of the cells.41, 42 Intriguingly, TIMP3 expression was increased in the first-trimester trophoblasts on hypoxic treatment.43 This implies that the increased expression of TIMP3 under hypoxic condition, a hallmark in preeclamptic trophoblast, may be mediated by the epigenetic alteration on its promoter.
Early detection of preeclampsia is necessary for effective treatment. We identified several genes with hypomethylation in their promoter regions. In particular, the significant reduction of DNA methylation in TIMP3 promoter of EOPET placentas could be useful as a biomarker for the disorder. Importantly, this site showed no significant change of DNA methylation by gestational age and there was a good intra-placental correlation in DNA methylation values. If further study shows that this methylation change is also conserved earlier in pregnancy, then measuring the DNA methylation level of TIMP3 in chorionic villus sampling (CVS) from pregnant women could reflect subsequent risk for EOPET.
Recent advances in measuring circulating fetal DNA from maternal plasma opens up an additional approach for non-invasive prenatal diagnosis.44 This strategy takes advantage of the fact that during pregnancy, 3 to 6% of cell-free DNA in maternal blood plasma is derived from the placenta.44 Therefore, one can detect abnormalities in the fetal DNA directly from the maternal blood without going through invasive methods such as amniocentesis and CVS. It has been shown that there is an over fivefold increase in circulating fetal DNA in the maternal plasma of preeclamptic pregnancies compared with their control counterparts as estimated by measuring the placental-specific unmethylated SERPINB5 DNA fragments.26 However, SERPINB5 is not differentially methylated between normal and preeclamptic placentas. The same extent of increase in circulating fetal DNA can also be found in preeclamptic maternal plasma by measuring SRY,45 suggesting that SERPINB5 is not a specific marker for preeclampsia. As TIMP3 is significantly hypomethylated in EOPET placentas the detection of an increased level of unmethylated TIMP3 cell-free DNA in the maternal plasma could provide increased sensitivity for the non-invasive screening of pregnancies for EOPET. Importantly, it possesses the same characteristics as SERPINB5 for being a potential universal non-invasive prenatal diagnostic marker: its methylation is specifically reduced in placenta but it is completely methylated in other tissues, including blood samples. SERPINA3, another gene in the SERPIN family, has been reported to be hypomethylated in severe preeclampsia, but the extent of methylation and its potential for being a clinical marker have not been examined thoroughly.8 We suggest, therefore, that the level of unmethylated TIMP3 DNA in maternal plasma could be a useful biomarker for early detection of severe preeclampsia.
In summary, we report the application of DNA methylation analysis to the elucidation of abnormal placental development associated with preeclampsia. Although DNA methylation at critical sites can reflect the availability of a gene for transcription, which may lead to altered expression depending on other regulatory factors present, it has a number of advantages over expression studies. First, it may be more resistant to the transient changes in gene expression associated with labor and delivery,12, 13, 14 as well as the effects of placental storage before sample processing.15 Although we did in this case observe an inverse association between TIMP3 methylation and expression, expression studies at term may not always reflect that which occurred during relevant periods of development. Second, the trend to hypomethylation of a variety of genes in EOPET suggests that loss of methylation may generally be involved in the response to hypoxia. Finally, DNA methylation differences provide an alternative approach for pre-symptomatic diagnosis of at risk pregnancies.
References
Roberts JM, Cooper DW : Pathogenesis and genetics of pre-eclampsia. Lancet 2001; 357: 53–56.
Redman CW, Sargent IL : Latest advances in understanding preeclampsia. Science 2005; 308: 1592–1594.
von Dadelszen P, Magee LA, Roberts JM : Subclassification of preeclampsia. Hypertens Pregnancy 2003; 22: 143–148.
Barker DJ : The long-term outcome of retarded fetal growth. Clin Obstet Gynecol 1997; 40: 853–863.
Wiles NJ, Peters TJ, Leon DA, Lewis G : Birth weight and psychological distress at age 45-51 years: results from the Aberdeen Children of the 1950 s cohort study. Br J Psychiatry 2005; 187: 21–28.
van Dijk M, Mulders J, Poutsma A et al: Maternal segregation of the Dutch preeclampsia locus at 10q22 with a new member of the winged helix gene family. Nat Genet 2005; 37: 514–519.
Kanayama N, Takahashi K, Matsuura T et al: Deficiency in p57Kip2 expression induces preeclampsia-like symptoms in mice. Mol Hum Reprod 2002; 8: 1129–1135.
Chelbi ST, Mondon F, Jammes H et al: Expressional and epigenetic alterations of placental serine protease inhibitors: SERPINA3 is a potential marker of preeclampsia. Hypertension 2007; 49: 76–83.
Nishizawa H, Pryor-Koishi K, Kato T, Kowa H, Kurahashi H, Udagawa Y : Microarray analysis of differentially expressed fetal genes in placental tissue derived from early and late onset severe pre-eclampsia. Placenta 2007; 28: 487–497.
Enquobahrie DA, Meller M, Rice K, Psaty BM, Siscovick DS, Williams MA : Differential placental gene expression in preeclampsia. Am J Obstet Gynecol 2008; 199: .
Founds SA, Conley YP, Lyons-Weiler JF, Jeyabalan A, Hogge WA, Conrad KP : Altered global gene expression in first trimester placentas of women destined to develop preeclampsia. Placenta 2009; 30: 15–24.
Torricelli M, Galleri L, Voltolini C et al: Changes of placental Kiss-1 mRNA expression and maternal/cord kisspeptin levels at preterm delivery. Reprod Sci 2008; 15: 779–784.
Torricelli M, Giovannelli A, Leucci E et al: Labor (term and preterm) is associated with changes in the placental mRNA expression of corticotrophin-releasing factor. Reprod Sci 2007; 14: 241–245.
Torricelli M, Giovannelli A, Leucci E et al: Placental neurokinin B mRNA expression increases at preterm labor. Placenta 2007; 28: 1020–1023.
Fajardy I, Moitrot E, Vambergue A, Vandersippe-Millot M, Deruelle P, Rousseaux J : Time course analysis of RNA stability in human placenta. BMC Mol Biol 2009; 10: 21.
Yuen RK, Avila L, Penaherrera MS et al: Human placental-specific epipolymorphism and its association with adverse pregnancy outcomes. PLoS One 2009; 4: e7389.
Robinson WP, Penaherrera MS, Jiang R et al: Assessing the role of placental trisomy in preeclampsia and intrauterine growth restriction. Prenat Diagn 2009; 30: 1–8.
Bourque DK, Avila L, Peñaherrera MS, von Dadelszen P, Robinson WP : Decreased placental methylation at the H19/IGF2 imprinting control region is associated with normotensive intrauterine growth restriction but not preeclampsia. Placenta 2010; 31: 197–202.
Audibert F, Friedman SA, Frangieh AY, Sibai BM : Clinical utility of strict diagnostic criteria for the HELLP (hemolysis, elevated liver enzymes, and low platelets) syndrome. Am J Obstet Gynecol 1996; 175: 460–464.
Douglas KA, Redman CW : Eclampsia in the United Kingdom. BMJ 1994; 309: 1395–1400.
Kramer MS, Platt RW, Wen SW et al: A new and improved population-based Canadian reference for birth weight for gestational age. Pediatrics 2001; 108: E35.
Tusher VG, Tibshirani R, Chu G : Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA 2001; 98: 5116–5121.
Zhang R, Zhou L, Li Q, Liu J, Yao W, Wan H : Up-regulation of two actin-associated proteins prompts pulmonary artery smooth muscle cell migration under hypoxia. Am J Respir Cell Mol Biol 2009; 41: 467–475.
Mayer W, Hemberger M, Frank HG et al: Expression of the imprinted genes MEST/Mest in human and murine placenta suggests a role in angiogenesis. Dev Dyn 2000; 217: 1–10.
Qi JH, Ebrahem Q, Moore N et al: A novel function for tissue inhibitor of metalloproteinases-3 (TIMP3): inhibition of angiogenesis by blockage of VEGF binding to VEGF receptor-2. Nat Med 2003; 9: 407–415.
Chim SS, Tong YK, Chiu RW et al: Detection of the placental epigenetic signature of the maspin gene in maternal plasma. Proc Natl Acad Sci USA 2005; 102: 14753–14758.
Chelbi ST, Vaiman D : Genetic and epigenetic factors contribute to the onset of preeclampsia. Mol Cell Endocrinol 2008; 282: 120–129.
Chiu RW, Chim SS, Wong IH et al: Hypermethylation of RASSF1A in human and rhesus placentas. Am J Pathol 2007; 170: 941–950.
Huppertz B : Placental origins of preeclampsia: challenging the current hypothesis. Hypertension 2008; 51: 970–975.
Oudejans CB, van Dijk M, Oosterkamp M, Lachmeijer A, Blankenstein MA : Genetics of preeclampsia: paradigm shifts. Hum Genet 2007; 120: 607–612.
Goswami D, Tannetta DS, Magee LA et al: Excess syncytiotrophoblast microparticle shedding is a feature of early-onset pre-eclampsia, but not normotensive intrauterine growth restriction. Placenta 2006; 27: 56–61.
Takahashi K, Kobayashi T, Kanayama N : p57(Kip2) regulates the proper development of labyrinthine and spongiotrophoblasts. Mol Hum Reprod 2000; 6: 1019–1025.
Winn VD, Haimov-Kochman R, Paquet AC et al: Gene expression profiling of the human maternal-fetal interface reveals dramatic changes between midgestation and term. Endocrinology 2007; 148: 1059–1079.
Feng H, Cheung AN, Xue WC et al: Down-regulation and promoter methylation of tissue inhibitor of metalloproteinase 3 in choriocarcinoma. Gynecol Oncol 2004; 94: 375–382.
Apte SS, Mattei MG, Olsen BR : Cloning of the cDNA encoding human tissue inhibitor of metalloproteinases-3 (TIMP-3) and mapping of the TIMP3 gene to chromosome 22. Genomics 1994; 19: 86–90.
Higuchi T, Kanzaki H, Nakayama H et al: Induction of tissue inhibitor of metalloproteinase 3 gene expression during in vitro decidualization of human endometrial stromal cells. Endocrinology 1995; 136: 4973–4981.
Montagnana M, Lippi G, Albiero A et al: Evaluation of metalloproteinases 2 and 9 and their inhibitors in physiologic and pre-eclamptic pregnancy. J Clin Lab Anal 2009; 23: 88–92.
Pang ZJ, Xing FQ : Expression profile of trophoblast invasion-associated genes in the pre-eclamptic placenta. Br J Biomed Sci 2003; 60: 97–101.
Xue WC, Chan KY, Feng HC et al: Promoter hypermethylation of multiple genes in hydatidiform mole and choriocarcinoma. J Mol Diagn 2004; 6: 326–334.
Noris M, Perico N, Remuzzi G : Mechanisms of disease: pre-eclampsia. Nat Clin Pract Nephrol 2005; 1: 98–114; quiz 120.
Shahrzad S, Bertrand K, Minhas K, Coomber BL : Induction of DNA hypomethylation by tumor hypoxia. Epigenetics 2007; 2: 119–125.
Gheorghe CP, Mohan S, Oberg KC, Longo LD : Gene expression patterns in the hypoxic murine placenta: a role in epigenesis? Reprod Sci 2007; 14: 223–233.
Koklanaris N, Nwachukwu JC, Huang SJ et al: First-trimester trophoblast cell model gene response to hypoxia. Am J Obstet Gynecol 2006; 194: 687–693.
Dennis Lo YM, Chiu RW : Prenatal diagnosis: progress through plasma nucleic acids. Nat Rev Genet 2007; 8: 71–77.
Lo YM, Leung TN, Tein MS et al: Quantitative abnormalities of fetal DNA in maternal serum in preeclampsia. Clin Chem 1999; 45: 184–188.
Acknowledgements
We would like to thank Ruby Jiang and Luana Avila for technical assistance, Alicia Murdoch, Jennifer Sloan and Lauren Currie for placenta recruitment, Dr Angela Devlin for use of the Biotage PyroMark MD and Dr Michael Kobor for use of the Illumina array. This work has been funded by a grant from the Canadian Institutes for Health Research (to WPR) and by a graduate student scholarship from the CIHR Interdisciplinary Women's Reproductive Health Training Program (to RKCY).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
RKCY is a co-inventor on a patent application for fetal methylation markers. Other authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Rights and permissions
About this article
Cite this article
Yuen, R., Peñaherrera, M., von Dadelszen, P. et al. DNA methylation profiling of human placentas reveals promoter hypomethylation of multiple genes in early-onset preeclampsia. Eur J Hum Genet 18, 1006–1012 (2010). https://doi.org/10.1038/ejhg.2010.63
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2010.63
Keywords
This article is cited by
-
The impact of DNA methylation as a factor of Adverse Pregnancy and Birth Outcomes (APBOs): a systematic review protocol
Systematic Reviews (2024)
-
Genomic imbalances in the placenta are associated with poor fetal growth
Molecular Medicine (2021)
-
Systematic review supports the role of DNA methylation in the pathophysiology of preeclampsia: a call for analytical and methodological standardization
Biology of Sex Differences (2020)
-
Effect of prematurity on genome wide methylation in the placenta
BMC Medical Genetics (2019)
-
No evidence for association of MTHFR 677C>T and 1298A>C variants with placental DNA methylation
Clinical Epigenetics (2018)
