Article Text

Abstract
Introduction Central lumbar spinal stenosis (LSS) is a common cause of pain, reduced function and quality of life in older adults. Current management of LSS includes surgery to decompress the spinal canal and alleviate symptoms. However, evidence supporting surgical decompression derives from unblinded randomised trials with high cross-over rates or cohort studies showing modest benefits. This protocol describes the design of the SUrgery for Spinal Stenosis (SUcceSS) trial —the first randomised placebo-controlled trial of decompressive surgery for symptomatic LSS.
Methods and analysis SUcceSS will be a prospectively registered, randomised placebo-controlled trial of decompressive spinal surgery. 160 eligible participants (80 participants/group) with symptomatic LSS will be randomised to either surgical spinal decompression or placebo surgical intervention. The placebo surgical intervention is identical to surgical decompression in all other ways with the exception of the removal of any bone or ligament. All participants and assessors will be blinded to treatment allocation. Outcomes will be assessed at baseline and at 3, 6, 12 and 24 months. The coprimary outcomes will be function measured with the Oswestry Disability Index and the proportion of participants who have meaningfully improved their walking capacity at 3 months postrandomisation. Secondary outcomes include back pain intensity, lower limb pain intensity, disability, quality of life, anxiety and depression, neurogenic claudication score, perceived recovery, treatment satisfaction, adverse events, reoperation rate and rehospitalisation rate. Those who decline to be randomised will be invited to participate in a parallel observational cohort. Data analysis will be blinded and by intention to treat. A trial-based cost-effectiveness analysis will determine the potential incremental cost per quality-adjusted life year gained.
Ethics and dissemination Ethics approval has been granted by the NSW Health (reference:17/247/POWH/601) and the Monash University (reference: 12371) Human Research Ethics Committees. Dissemination of results will be via journal articles and presentations at national and international conferences.
Trial registration number ACTRN12617000884303; Pre-results.
- placebo controlled trial
- randomised controlled trial
- lumbar spinal stenosis
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
This will be the first randomised placebo-controlled trial of surgery for symptomatic lumbar spinal stenosis (LSS).
Participants and study assessors will be blinded to treatment allocation.
The placebo intervention will resemble surgical decompression, but exclude removal of the bone and ligament (ie, to increase the diameter of the spinal canal).
The findings of this study will provide level 1 evidence for the treatment efficacy, safety and cost-effectiveness of decompression for LSS.
Introduction
Lumbar spinal stenosis (LSS) is a common source of pain and reduced function in those over the age of 50.1 The condition is attributed to a reduction in the diameter of the lumbar spinal canal from age-related degenerative changes of the surrounding structures, including the intervertebral discs, facet joints and ligaments.2–4 Symptoms typically include neurogenic claudication, which is defined as pain, paraesthesia and/or fatigue in the gluteal area and/or legs that is aggravated by walking, and relieved by bending forward or sitting.5
In the Japanese population, approximately three-quarters of those over 40 years of age have MRI signs of moderate central canal stenosis (ie, narrowing by one-third of the canal area).3 Thirty per cent have severe stenosis (narrowing by two-thirds), but only around one-fifth of those report clinical symptoms.3 A case-multiple control study from the USA including 126 individuals (50 with a clinical diagnosis of lumbar stenosis, 44 with back pain and no diagnosis of stenosis and 32 with no pain) found no correlation between imaging findings and clinical diagnosis of stenosis, with approximately 50% (n=13/32) of the asymptomatic participants being diagnosed with stenosis by blinded assessors.6 The diagnosis of LSS, therefore, requires both the presence of clinical symptoms (ie, neurogenic claudication) and evidence of stenosis on diagnostic imaging (eg, MRI).3 Current guidelines recommend symptomatic LSS be managed initially with non-operative treatment,7 including oral medication (ie, non-steroidal anti-inflammatory drugs and analgesics) and physical therapies (eg, exercise therapy or manual therapy).8 If patients with symptomatic LSS fail to improve with non-operative treatment, referral for surgical assessment is recommended.7 9
Surgical management of LSS involves decompression of the spinal canal via laminectomy or laminotomy.10–12 The procedure is the most common form of spinal surgery in adults over the age of 65.13 Recent data from the USA estimate an adjusted rate of lumbar stenosis surgery of 135.5 per 100 000 Medicare beneficiaries, with a resulting aggregated hospital bill of US$1.65 billion per year. The evidence supporting this practice is, however, inconclusive. The latest Cochrane review assessing the efficacy of surgery compared with non-surgical care for this population included five randomised controlled trials (643 participants) and concluded there is low quality evidence that surgery provides little benefit after 2 years follow-up compared with non-surgical treatment (a difference in the Oswestry Disability Index (ODI) of 4.43, which is less than the accepted minimally clinically important difference of 15).14 This is in part attributed to the methodological limitations of existing trials, including lack of participant and assessor blinding, imprecise results due to small samples and high treatment crossovers.8 15–18 For example, the Spine Patient Outcomes Research Trial (SPORT) published in 2008 evaluated the effects of surgery for spinal canal stenosis19 in an open randomised parallel group design. Over one-third of participants assigned to surgery did not undergo surgery, and almost half (43%) of those assigned to non-operative management underwent surgery. Given patients often have strong preferences for one treatment over the other, treatment crossovers are common in pragmatic open trials of surgical interventions.19 20 The results of the as-treated analysis in the SPORT trial favoured surgery in reducing pain and disability, but these results were not confirmed in the intention-to-treat analyses. A more recent trial by Delitto et al that randomised 169 patients to either decompressive surgery or physical therapy20 also failed to find any clinically important differences in pain improvement or physical function between treatment groups. Given the lack of robust evidence confirming the superiority of surgery compared with non-surgical care; the benefits of surgery observed in clinical practice might be attributable to non-specific effects, including regression to the mean and the placebo effects associated with invasive procedures.21 These findings highlight the need for a placebo-controlled trial of surgical decompression in this population.
The importance of a placebo surgical intervention is that it is indistinguishable (ie, to the participant) from the traditional surgical procedure in many aspects. These include preoperative and anaesthetic management; incision length and surgical dissection; and postoperative discomfort and management. By design and intention, the observable differences between participants receiving placebo surgical intervention and those receiving the traditional surgical procedure are more reliably due to the procedure itself and not the events surrounding the procedure. In the case of lumbar surgical decompression, this means that the placebo surgical intervention must include muscle dissection down to the bone of the lumbar lamina, but no removal of bone or ligamentum flavum. Placebo interventions also differ from sham in that a sham intervention might only involve a skin incision; falling short of replicating the experience of the traditional surgical procedure, including the absence of postsurgical pain from muscle dissection, or any potential benefits from denervation effects of surgical dissection.
Placebo-controlled trials of surgical interventions are not new. In the late 1950s, two landmark randomised trials for patients with severe angina pectoris compared the common practice of internal mammary artery ligation to placebo surgical intervention.22 23 The observation that the placebo surgical intervention resulted in similar and sustained symptom relief as true ligation surgery disproved the rationale of the procedure to increase coronary artery blood flow. A recent systematic review identified a total of 53 randomised placebo-controlled trials of surgery published between 1946 and 2013, with about half showing the index procedure was superior to a placebo comparator, while the other half showed no superiority of the surgical procedure.24 The review also found that placebo trials can be safe, and that placebo interventions are, in general, associated with less frequent and less severe adverse events than the experimental group.
Given the growing rates of surgical management of symptomatic LSS and the inconclusive evidence supporting its efficacy, we have designed and will conduct the first placebo-controlled trial of surgical decompression for this population. The aim of the SUrgery for Spinal Stenosis (SUcceSS) trial is to evaluate the efficacy, safety and cost-effectiveness of decompressive surgery for people with severe LSS who have failed to respond to non-operative care. We will evaluate relevant participant outcomes related to LSS, including disability, walking capacity, pain, quality of life, serious adverse events (SAEs) and reoperation rates. Our study hypothesis is that decompression surgery is more effective, more cost-effective and safer than placebo surgical intervention in improving pain and function in patients with LSS. The study will also include a parallel observational study of eligible participants who decline to be randomised to the placebo-controlled trial to test for any selection bias.
Methods
Study design
This paper describes a research protocol for the SUcceSS trial, a prospectively registered, randomised placebo-controlled trial of decompressive spinal surgery for LSS. The participants, investigators (other than the surgical team performing the procedures) and outcome assessors will be blinded to treatment allocation. The protocol was developed in accordance with Standard Protocol Items: Recommendations for Interventional Trials and Template for Intervention Description and Replication statements.25 26 The study design appears in figure 1.
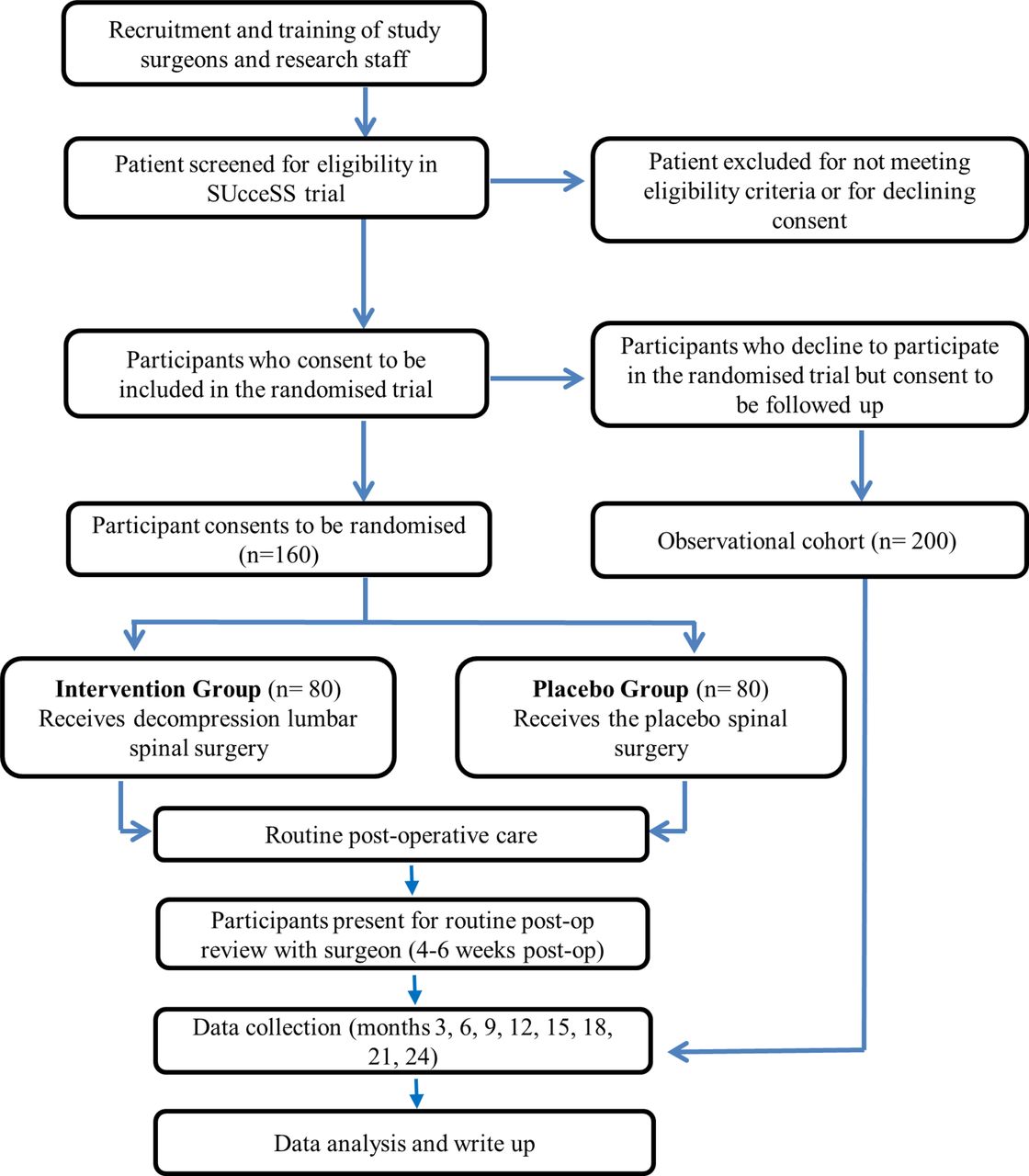
{kind=link}
The flow chart of the SUcceSS study design. SUcceSS, SUrgery for Spinal Stenosis.
Participants and recruitment
Consecutive patients who present to one of the study surgeons in New South Wales or Victoria, Australia will be assessed for eligibility and invited to participate. All study surgeons will be qualified orthopaedic spine surgeons or neurosurgeons, and are required to have current registration to perform spinal decompression surgery in Australia.
Inclusion criteria
Participants will need to meet all the following inclusion criteria:
Be 50 years of age or older.
Present with neurogenic claudication for at least 3 months. Neurogenic claudication is defined as pain, numbness and/or fatigue below the gluteal line with or without back pain (if back pain is present, leg pain is greater than back pain) that is precipitated by walking and alleviated by sitting or other posture of lumbar flexion.
Have grades C or D stenosis as defined by Schizas et al 27 indicating occlusion (absent cerebrospinal fluid signal) of the central lumbar spinal canal at one or two levels on T2-weighted MRI or CT myelogram.
Be considered by a study surgeon to be in need of single or dual-level decompressive surgery.
Have not improved with non-surgical treatment (eg, physiotherapy, medication).
Exclusion criteria
Participants will be excluded if they meet any of the following criteria:
Are pregnant.
Are under a worker’s compensation claim.
Have been diagnosed with serious spinal pathology including: cancer, infection, cauda equina syndrome, spinal fracture.
Present with active Paget’s disease of the spine.
Have a diagnosis of lumbar instability defined as more than 4 mm translation or 10° of angular motion between flexion and extension on upright lateral radiographs (to exclude participants who might need to undergo concurrent surgical fusion).
Have had previous lumbar spine surgery at the same levels.
Inadequate English to complete outcome measures.
Motor deficit related to lumbar compression (Medical Research Council grades 0–4) and the motor deficit interferes with walking ability.
Presence of significant scoliosis (Cobb angle >25°) or other spinal deformities.
Presence of known or demonstrated peripheral vascular disease causing vascular claudication, that is, claudication accompanied by absent foot pulse or vascular insufficiency detected with Doppler Ultrasound or CT angiography.
Meyerding classification grade 2 or greater spondylolisthesis.
Symptomatic hip disease with symptoms reproduced with external or internal rotation of the hip joint.
Cognitive impairment interfering with participant’s ability to give full and informed consent or complete the baseline or follow-up assessments.
If a study surgeon determines the participant has met the trial eligibility criteria, the participant will be provided with information about the trial and asked if they consent for a study researcher to contact them. As part of consent, participants will be informed they could be allocated on a 50:50 basis to receive either conventional surgical decompression or placebo surgical intervention (no decompression). A researcher will then contact them, provide detailed trial information, obtain written informed consent and collect all baseline assessments. A subject/participant identification (ID) number will then be allocated and the participant booked for surgery at the earliest available date. When surgery cannot be scheduled within 6 weeks from baseline, baseline assessments will be repeated to ensure a maximum of 6 weeks from baseline assessment to randomisation. Eligible patients who do not consent to participate in the randomised trial will be invited to participate in a parallel observational cohort. Participants who consent to participate in the observational cohort will be followed up for the duration of the trial and the same outcome measures will be collected at all time points.
Study treatment
On the scheduled day of surgery, each participant will be admitted to the hospital of the recruiting study surgeon and follow the hospital’s routine admission protocol. Participants will receive general anaesthesia, venous thromboembolism prophylaxis and prophylactic antibiotics. Based on each individual surgeon’s standard surgical practice, a local anaesthetic may be infiltrated into the subcutaneous tissues, with a single midline or dual paramedian longitudinal skin incision made through the skin and adipose layers. The posterior spinal muscles will then be dissected in a subperiosteal fashion to expose lumbar laminae and self–retaining retractors placed. Participants will be randomised to treatment groups following muscle dissection. If the patient is randomised to placebo surgical intervention, the surgeon will progress no further and close the wound. In this case, the lumbar spinal canal will not be entered or the dural sac decompressed. If the patient is randomised to receive surgery involving decompression, the surgeon will proceed to remove bone from the lamina, and underlying ligamentum flavum in order to enter the lumbar spinal canal and decompress the dural sac. Following the decompression or placebo surgical intervention, routine postoperative care will follow as per the standard of care from the operating hospital (eg, mobility advice, wound care).
Randomisation
Central randomisation will be used to ensure concealment of treatment allocation. An interactive voice response will be used for automatic, 24 hours/day, 7 days/week allocation of participants and delivered to participating surgeons at the time of surgery. The randomisation will be obtained by a study trained theatre clinician who will contact the central randomisation service following muscle dissection, but prior to decompression. Randomisation will be by random permuted blocks of 4 and 6 and stratified by surgeon.
Blinding
Participants, assessors and investigators (other than the treating surgical team) will be blinded to treatment allocation. Randomisation will occur in the surgical theatre to eliminate treatment bias by any of the surgical team prior to the procedure. Members of the surgical team will not be involved in patient care after the procedure. Arrangements will be made for surgical colleagues who are unaware of the treatment given to provide postoperative care.
Data collection
Blinded research staff will collect all baseline data including demographic data (eg, work status, socioeconomic status, symptom duration, etc) and outcome data, SAEs, and hospital admissions and medications. Outcomes will be collected at baseline and at 3, 6, 12 and 24 months (with adverse events and hospital admissions additionally collected at 9, 15, 18 and 21 months). All research staff will be trained to ensure data accuracy, consistency and completeness.
Primary outcomes
The coprimary outcomes will be the function measured with the ODI score28; and the proportion of participants who have meaningfully improved their walking capacity.29 Our selection of coprimary outcomes reflects the two main complaints of patients with LSS: walking capacity and function. The ODI is a self-reported questionnaire commonly used to assess function in people with spinal conditions, including LSS.19 20 30 The outcome has been validated in this population, showing good postsurgery responsiveness, excellent internal consistency (Cronbach’s alpha of 0.9) and test–retest reliability (Intra-class correlation (ICC): 0.89).31 The ODI is also strongly correlated with patient satisfaction after surgery.31
The self-reported walking capacity measure has also been validated in people with LSS and neurogenic claudication, showing strong correlation with the ODI walking section (Spearman r: 0.64) and self-paced walking test (Spearman r: 0.65; ICC: 0.68).32 33 Meaningful improvement will be defined as a score of 6 or 7 on the walking capacity change scale: ‘How would you say your walking capacity is today compared with immediately before surgery?’, where 1 is ‘a great deal worse’ and 7 is ‘a great deal better’.29 Coprimary outcomes will be measured at baseline (ODI only), 3, 6, 12 and 24 months. The primary outcome time point will be 3 months because the beneficial effects of definitive surgical decompression are likely to be apparent by then, and patients with poor clinical response are more likely to undergo further spinal imaging to assess the adequacy of decompression, resulting in unblinding.
Secondary outcomes
Secondary outcomes include:
Walking ability, using the walking section of the ODI,29 will be measured at baseline, 3, 6, 12 and 24 months.
Assessment of neurogenic claudication: claudication will be measured using the Swiss Spinal Stenosis Questionnaire31 and will be measured at baseline, 3, 6, 12 and 24 months. Results of the symptom severity and functional subscales of the Swiss Spinal Stenosis Questionnaire will be separately reported.
Average lower limb pain in the past week will be measured using the 11-point Numerical Rating Scale (NRS), where 0 is ‘no pain’ and 10 is ‘worst possible pain’,34 35 at baseline, 3, 6, 12 and 24 months.
Average low back pain in the past week will be measured using the 11-point NRS scale, where 0 is ‘no pain’ and 10 is ‘worst possible pain’,34 35 at baseline, 3, 6, 12 and 24 months.
Quality of life: the Assessment of Quality of Life questionnaire will be used to assess the quality of life and to estimate quality-adjusted life years (QALYs) for the cost-effectiveness analyses.36 37 It will be measured at baseline, 3, 6, 12 and 24 months.
Self-reported perceived recovery will be measured using a 7-point Likert scale where 1 is ‘worse than ever’ and 7 is ‘completely recovered’38 will be measured at 3, 6, 12, and 24 months.
Satisfaction with surgery: measured using section XIII of the Swiss Spinal Stenosis Questionnaire, which asks ‘how satisfied are you with the overall result of your back operation’ where 1 is very satisfied and 4 is very dissatisfied31 will be measured at 3 months only.
Reoperation and rehospitalisation rates: data on hospital and/or privately funded healthcare visits will be collected over the phone via the use of a diary which the participant will keep and complete at all listed time points. These data will be measured at 3, 6, 9, 12, 15, 18, 21 and 24 months.19
Healthcare utilisation will be collected from participant diaries for the cost-effectiveness analysis at 3, 6, 9, 12, 15, 18, 21 and 24 months.
Adverse events will be collected from participant diaries at screening, baseline, 3, 6, 9, 12, 15, 18, 21 and 24 months following treatment.
Surgical decompression and placebo intervention fidelity: an MRI scan will be performed on a random 20% selection of those receiving the surgical decompression or placebo interventions to assess the fidelity of the two procedures. This will be completed at the 24-month follow-up only.
Other data collected
Demographic information, clinical data and expectation of treatment outcome will be collected at baseline. These measures include an expectation of satisfaction with treatment outcome in terms of pain and walking capacity measured as: ‘How much pain relief would you expect from treatment?’ and ‘How much improvement in your walking capacity would you expect from treatment?’. Participants will be asked to rate their expectation of change in walking capacity and expected pain relief using a 7-point Likert scale where 1 is ‘no improvement expected’ and 7 is ‘full recovery expected’.39 Anxiety and depression will be measured at baseline via the Hospital Anxiety and Depression Scale.40 Blinding fidelity will be assessed by asking the patient ‘which study group do you think you were in?’ (decompression surgery, placebo intervention, do not know) and will be measured at hospital discharge following surgery and at 3 months.
Adverse events
A standard operating procedure (SOP) for clinical trial safety will be used to guide the data collection of adverse events, SAEs, suspected and unexpected serious adverse reactions in this study. This SOP will describe in detail the actions to be taken should adverse events occur and the relevant timelines. Events will be classified according to their attribution (not related, doubtful, possible, probable and very likely) and severity (mild, moderate or severe). Risks and complications of surgical decompression are rare and may include cerebrospinal fluid leak, postoperative instability of the operated level, infection, nerve root damage and bleeding.
SAEs are defined as any event that is life threatening, results in death, hospitalisation or significant disability.19 Any SAE will be immediately reported to the data monitoring committee by a notified study member. The steering committee will investigate the nature of the adverse event and whether unblinding of treatment allocation is necessary. The ethics committee will also be informed of all SAEs.
Treatment fidelity
All staff involved in the delivery of the study will be required to undertake research training. This will include study surgeons, anaesthetists and nurses involved in the care of participants. Regular research meetings will be conducted with study staff, and site visits of participating hospitals completed. Study patients will be provided with a study pamphlet that can be shown to their other treating healthcare professionals (eg, physiotherapists, general practitioners, etc). The pamphlet explains the trial and their participation in it.
Unblinding
Unblinding will be carried out in the case of SAEs where knowledge of the participant’s treatment allocation is necessary for further medical management of the participant, or in cases where the participant requests to be unblinded due to his/her withdrawal from the study. Following unblinding, the following information must be provided: the reason for unblinding, details of the clinician, the date and time the decision was made, and any supporting documentation. Regardless of unblinding, a follow-up schedule for data collection will be attempted, to enable a full analysis of all participant data on an intention-to-treat basis. At the end of the trial, participants may be notified of the study results and treatment allocation if they wish to be.
Data integrity
A data monitoring committee has been convened and will overview data collection and integrity. Data collected by the trained research staff will be directly entered into a custom-built Electronic Data Capture program at the time of data collection, with a prompt for double checking of the accuracy of the primary outcomes. Any inconsistencies in the data will be explored and resolved. Any data completed by participants via questionnaires will be recorded into the database by the research assistant.
A database will be backed-up regularly on a secure network and compliant to the Note for Guidance on Good Clinical Practice, according to our data management plan. Study personnel will only be able to access the database with a personal login and password.
Retention of documents
The study investigators will maintain adequate and accurate records to enable the conduct of the study to be fully documented and the study data to be subsequently verified. These documents will be classified into two separate categories (1) investigator’s study file and (2) participant clinical source documents. The investigator’s study file will contain the protocol/amendments, schedule of assessments, independent ethics committee/institutional review board and governmental approval with correspondence, sample informed consent, staff curriculum vitae and authorisation forms and other appropriate documents/correspondence.
Should the study investigators wish to assign the study records to another party or move them to another location, the sponsor must be notified in advance. After the completion of the study, study data will be archived by the sponsor for a minimum of 15 years.
Statistical analysis
Treatment effectiveness analyses of randomised trial data will be blinded and performed on an intention-to-treat basis. Statistical significance will be defined as p<0.025 on the basis of a two-sided test (Bonferroni correction for co-primary outcomes). The 3-month follow-up will be the primary endpoint. All between-group differences at all follow-up time points will be analysed with linear regression for continuous outcomes and logistic regression for dichotomous outcomes. Adjusted (sensitivity) and unadjusted (main) analyses will be presented for the main confounders. Heterogeneity between recruiting hospitals will be calculated and adjusted for in sensitivity analyses by including hospital as a covariate in the models. To elucidate if any sampling bias is present in the trial, baseline characteristics of participants in the randomised cohort will be contrasted with those in the observational cohort.
In addition to a primary and secondary outcome analyses, a formal interim analysis will be conducted after two-thirds of participants (n=107) have completed the 3-month follow-up of the coprimary outcome measurements (primary endpoint). The interim analysis will be performed by a blinded statistician who can recommend to the data monitoring committee that the trial is terminated early for efficacy, defined as a between-group difference greater than 3 SD. A recommendation to terminate the study early could also be made if there is proof beyond reasonable doubt that the intervention or placebo cause an unacceptable net harm. The trial will not be terminated on the grounds of futility.
Sample size calculation was based on the between-group difference at all follow-ups, on coprimary outcomes. A sample size of 80 per group (total of 160 participants) will achieve 90% power to detect a minimum clinically important difference of 15 points (of a total of 100 points) on ODI (SD: 18) and difference between groups in proportion of participants who have improved in the walking change score (ie, 6 or 7 on the Likert scale) of 30% (ie, assuming 30% of participants in the placebo group and 60% in the intervention group will have improved).29 This sample size allows for a 15% loss to follow-up rate; 5% crossover between groups at 3 months19 20 41 and provides enough power to detect an absolute difference of 20% in reoperation rates over 2 years (the current reoperation rate for decompressive surgery is 7% and the rate of surgery in the non-operative groups of previous studies is approximately 50%).42
Cost-effectiveness analysis
In the event of an observed positive treatment effect, a cost-effectiveness analysis will be conducted. Intervention costs (staff, consumables, equipment, etc) will be ascertained from financial statements from participating sites. Hospital admissions over the course of the study in both patient arms will be recorded in patient diaries and costed on the basis of published diagnosis-related group cost weights. Non-hospital services will be costed through individually linked Medicare data; costs of non-hospital medications will be determined through individually linked Pharmaceutical Benefits Scheme data. The aggregate of relevant costs to patients in both arms will be used to calculate the incremental healthcare costs incurred (or cost savings). QALYs will be used to determine effectiveness, and converted into a utility index, using data derived from the Australian population. Average differences in QALYs will be estimated between treatment and control arms. An incremental cost-effectiveness ratio will be calculated based on the ratio of incremental costs over incremental QALYs. Sensitivity analyses will be conducted to determine the robustness of the results to key assumptions made in the analyses.
Patient and public involvement
Consumer representatives were involved in different stages of the design of this trial. Prior to the protocol development phase, patients presenting to neurosurgeons or orthopaedic surgeons with LSS and indication for surgical decompression were surveyed regarding their views on the value of such a study and their willingness to participate in this trial. Members of the Consumer Advisory Group of the Australia and New Zealand Musculoskeletal (ANZMUSC) Clinical Trials Network were also consulted and asked to provide feedback on the protocol during its development phase.
Ethics and dissemination
The SucceSS trial will be undertaken across multiple sites in New South Wales and Victoria, Australia and a report of the trial findings will be prepared according to the consort Consolidated Standards of Reporting Trials statement. Authorship eligibility guidelines of publications arising from the SUcceSS study will follow those outlined by the International Committee of Medical Journal Editors (http://www.icmje.org/). Findings from the study will be disseminated via journal publications and presentations at national and international conferences. The study is sponsored by The University of Sydney, Australia and centrally coordinated and managed by staff based at the Kolling Institute/Northern Clinical School. The sponsor has no role in the design of the trial. A Steering Commitee is responsible for study conception, design, protocol refinement and providing the scientific direction of the study. Members of the committee have expertise in the conduct of large, high-quality randomised trials, surgical trials and placebo trials of surgery. The current protocol is V.3.0 (13/07/2018). Any modifications to the protocol, which may impact the study design and conduct, potential benefit or harm of the participants, will require a formal amendment to the protocol. Such amendment will be agreed on by the Steering Commitee and approved by the ethics committee prior to implementation. The study adheres with the Australian National Health and Medical Research Council ethical guidelines for human research.
Discussion
This manuscript outlines the design of SUcceSS, the first randomised placebo-controlled trial of surgery for LSS. The trial has been endorsed by the ANZMUSC Clinical Trials network; reflecting its robust design. The design of the SUcceSS trial is the result of an ongoing and close collaboration among researchers, health economists, biostatisticians, consumers and surgeons. SUcceSS will address the main limitations of previous surgical trials via its unique study design and use of a placebo arm. The inclusion of a placebo surgical intervention will ensure blinding of participants and assessors to treatment allocation, limiting treatment crossover and account for any placebo effect associated with decompression surgery. As a result, the study will provide high-quality evidence for the efficacy of decompressive surgery in treating LSS.
Our trial conforms to the ethical framework for the use of placebo procedures in clinical trials proposed by Horng and Miller.43 According to the framework, the conduct of a placebo-controlled trial is ethical when: (1) it involves an important research question that cannot be answered without a placebo control. This is the case in surgical trials of LSS, given most commonly used outcome measures are self-reported and blinding of participants is impossible in ‘open-label’ trials or those involving a no-treatment control arm. In this case, there is no alternative to placebo and no other sufficiently rigorous trial design that has less risk (any other option will affect blinding); (2) although the risks are greater than minimal they are likely to be equal in both groups. Past research has shown that the risk associated with placebo surgery is not greater than that of active surgery. Moreover, there are no anticipated extra risks and hazards to patients allocated to the placebo surgical intervention group, since there will be no bone removal24; (3) risks associated with the placebo and active interventions do not exceed a threshold of acceptable research risk. We have been working closely with an ethicist to ensure all potential risks involved with participating in any of the interventions are appropriately disclosed to the participant; (4) any risk in either arm is minimised by involving highly experienced and trained surgeons, following a carefully designed protocol and establishing a data monitoring committee and (5) informed consent is sought prior to any study-related procedures being conducted.
One debate is whether the comparator for full surgical treatment should be placebo or sham surgery. Placebo surgical intervention is a surgical intervention performed in exactly the same way as the procedure under investigation, but omitting the critical surgical element or mechanism, given the mechanism by which it potentially obtains benefit has been questioned.44 Placebo is designed to cause the same effects on the participant as definitive surgical treatment (such as discomfort), except for the critical element (canal decompression). It differs from a sham intervention which has only superficial similarity with definitive surgery, such as skin incision only. Placebo surgical intervention is frequently associated with larger beneficial effects on the participant than a sham. Furthermore, placebo surgical intervention also allows for more reliable blinding of patients and assessors, than any other comparator (ie, no treatment, non-surgical care and sham). Therefore, to ensure a more robust design and given we currently lack strong evidence supporting the therapeutic mechanism of decompression surgery (ie, widening of the spinal canal), the SucceSS trial team have opted to include a placebo surgical intervention.
References
Footnotes
Patient consent for publication Not required.
Contributors MF, IH, RB and CM had the original idea. MF, IH, GAD, RS, DB, QL, SJ, RJM, CM, JL and RB were involved in the study design and secured funding. DBA, MF, IH, GAD, RS, DB, QL, SJ, RJM, CM, RY, TZ, JL and RB were involved in the protocol development. DBA and MF wrote the first draft. All authors have approved the final draft.
Funding This work is funded by the National Health and Medical Research Council (NHMRC) Project Grant (APP1125140). MF is funded by an NHMRC Career Development Fellowship (APP1143593) and a Sydney Medical Foundation Fellowship. SJ is funded by an NHMRC Principal Research Fellowship. CM is funded by an NHMRC Principal Research Fellowship (APP1103022). RB is funded by an NHMRC Senior Principal Research Fellowship (APP1082138).
Competing interests None declared.
Ethics approval Ethics approval has been granted by the South Eastern Sydney Local Health District Human Research Ethics Committee (reference: 17/247/POWH/601) and the Monash University Human Research Ethics Committee (reference: 12371).
Provenance and peer review Not commissioned; externally peer reviewed.