Article Text

Abstract
Objectives Heterozygous familial hypercholesterolaemia (FH) confers a significant risk for premature cardiovascular disease (CVD). However, the estimated prevalence of FH varies substantially among studies. We aimed to provide a summary estimate of FH prevalence in the general population and assess variations in frequency across different sociodemographic characteristics.
Setting, participants and outcome measures We searched MEDLINE, EMBASE, Global Health, the Cochrane Library, PsycINFO and PubMed for peer-reviewed literature using validated strategies. Results were limited to studies published in English between January 1990 and January 2017. Studies were eligible if they determined FH prevalence using clinical criteria or DNA-based analyses. We determined a pooled point prevalence of FH in adults and children and assessed the variation of the pooled frequency by age, sex, geographical location, diagnostic method, study quality and year of publication. Estimates were pooled using random-effects meta-analysis. Differences by study-level characteristics were investigated through subgroups, meta-regression and sensitivity analyses.
Results The pooled prevalence of FH from 19 studies including 2 458 456 unique individuals was 0.40% (95% CI 0.29% to 0.52%) which corresponds to a frequency of 1 in 250 individuals. FH prevalence was found to vary by age and geographical location but not by any other covariates. Results were consistent in sensitivity analyses.
Conclusions Our systematic review suggests that FH is a common disorder, affecting 1 in 250 individuals. These findings underscore the need for early detection and management to decrease CVD risk.
- familial hypercholesterolemia
- prevalence
- frequency
- systematic review
- meta-analysis
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Strengths and limitations of this study
Use of an extensive search strategy and adherence to predetermined inclusion/exclusion criteria.
Use of evidence-based inverse variance weighted random effects meta-analysis to quantify a robust estimate of the pooled frequency of heterozygous familial hypercholesterolaemia in adults.
Our study possesses a large sample size (n=2 458 456).
We include only English-language peer-reviewed studies making it possible that some relevant articles were not included.
Our analyses possessed considerable amount of quantifiable heterogeneity.
Background
The frequency of heterozygous familial hypercholesterolaemia (FH) was originally reported as 1 in 500 (0.2%).1 This estimate is based on work that determined the prevalence in homozygous individuals and used Hardy-Weinberg principles to calculate the frequency in heterozygotes.2 Similar frequencies have been described in subsequent reports of population-based samples.3–7 However, this estimate has recently been criticised for its imprecision.8 Human behaviour does not adhere to Hardy-Weinberg assumptions (eg, random mating, no migration) and violations of these principles have been shown to significantly impact the results of gene-disease association studies.9 Further, recent work indicates as many as 1 in 200 people may be affected by FH10–12 and there are some data to suggest that regional variations in FH frequency exist.13–19
The population prevalence of FH is difficult to determine for several reasons. Most countries lack national FH registers or large observational databases. Yet, even when such databases exist, they often contain insufficient data on aspects of clinical histories essential for FH diagnosis. No uniform criteria for FH diagnosis exist and the three sets of criteria commonly used vary in the amount of emphasis placed on clinical characteristics in determining FH. Additionally, the ability to detect such findings may vary based on the clinical acumen and experiences of assessors.20 Genetic diagnosis has the potential to mitigate confounding inherent in clinical diagnostic criteria. However, the feasibility and cost-effectiveness of genetic screening continues to be debated,8 21–23 a high proportion of patients with clinical FH diagnoses may not be identified24 and all of the genetic mutations that cause FH may not yet be known. Together, these factors suggest the potential for a different FH frequency than original estimates.
Ascertaining the prevalence of FH has important clinical and public health implications, especially in light of the availability of new but expensive treatments (eg, proprotein convertase subtilisin/kexin type 9 (PCKS9) inhibitors) for this condition. FH is caused by defects in the low-density lipoprotein receptor (LDLR) pathway, resulting in elevated LDL-cholesterol (LDL-C) concentrations that are largely resistant to caloric restriction, weight loss and physical exercise interventions in affected individuals.24 FH also predicts a very high risk of cardiovascular disease (CVD) even in the absence of other traditional risk factors as patients possess these LDL-C concentrations from birth.25 Early diagnosis and treatment of FH with lipid-lowering therapy has proven to be both cost efficient and effective in mitigating cardiovascular morbidity and mortality risk.26 27 Despite these benefits, numerous reports suggest that FH is currently underdiagnosed in the general population27 and that in some jurisdictions, a large proportion of affected individuals have difficulty accessing effective lipid-lowering therapies.28 Clinicians routinely consider estimates of disease prevalence, variations in different population groups (eg, age, sex, ethnicity) and the presence of known risk factors in formulating differential diagnoses. These factors also form important considerations when evaluating national strategies for the optimal identification and treatment of individuals.29 Thus, determining the prevalence of FH and its variation by sociodemographic factors provides an important first step in reducing disease burden.
While a number of narrative and systematic reviews have summarised studies of FH,8 13 30–34 there has been no attempt to consolidate these studies to derive a robust prevalence estimate or to assess variation according to sociodemographic factors. We therefore aimed to systematically review the existing literature presenting estimates of FH in the adult general population and explore variation in prevalence estimates by age, sex, geographical location and study quality.
Methods
We carried out a systematic review and meta-analysis in accordance with the Meta-analysis Of Observational Studies in Epidemiology consensus statement.35 The protocol for this review was registered with the PROSPERO International Prospective Register of Systematic Reviews (CRD42016042208).
Study identification and selection
This study was part of a series of systematic reviews with a standardised search strategy examining the disease burden posed by heterozygous FH. We searched MEDLINE, EMBASE, PsycINFO, Global Health, the Cochrane Library and Pubmed (for publications ahead of print) for published, peer-reviewed literature using controlled vocabulary and keywords related to FH and relevant epidemiological terms. Results were limited to human studies published in English between 1 January 1990 and 31 January 2017. We reviewed reference lists of all included articles and relevant literature reviews, systematic reviews and meta-analyses for additional eligible studies. A detailed search strategy is included in the supplement to this manuscript (see online supplementary etable 1).
Supplementary file 1
Titles and abstracts and full texts were evaluated in duplicate by independent reviewers (LEA, SDS) using standardised forms (see online supplementary etable 2). Disagreements were resolved through discussion to consensus. For inclusion in the systematic review of prevalence, studies were required to include live human participants and to report on the prevalence of FH. Studies were included if they ascertained FH frequency using one of the following methods (see online supplementary etables 3–5): (1) DNA-based evidence of LDLR, apolipoprotein-B (Apo B), or PCSK9 mutations; (2) Dutch Lipid Clinic Network (DLCN) criteria; (3) Simon Broome Registry (SBR) criteria; (4) Making Early Diagnosis to Prevent Early Death (MEDPED) criteria or (5) total cholesterol levels (>290 mg/dL or 7.5 mmol/L) or LDL-C levels (>189 mg/dL or 4.9 mmol/L).34 We did not include articles reporting on the prevalence of or regional variations in specific LDLR, Apo B or PCSK9 mutations in study populations given their potential to underestimate FH frequencies.
Data extraction
One reviewer (LEA) independently extracted data regarding study characteristics (eg, design, population characteristics, diagnostic measures, prevalence estimates) from the full text of included articles. Another reviewer (RLR) checked the extracted data and any detected discrepancies were resolved. We did not attempt to contact authors of studies with missing or incomplete data nor did we exclude any such studies from our synthesis.
Study quality assessment
Two reviewers (LEA, RLR) independently assessed the quality of eligible studies using the Effective Public Health Practice Project (EPHPP) Quality Assessment Tool for Quantitative Studies (http://www.ephpp.ca/tools.html) and resolved discrepancies through consensus. It has been shown to be acceptable for use in evaluating a variety of study designs including randomised controlled trials, before-and-after studies and case control studies (see online supplementary etable 6). The tool assesses study quality across six domains: selection bias; study design; confounding variables; blinding protocols; data collection methods and handling of withdrawals and dropouts. Each dimension is rated on a three-point scale—strong, moderate and weak—and these ratings feed into a global rating of study quality. Global study quality is considered to be strong if none of the quality domains is rated as weak, moderate if one domain is rated as weak and weak if two or more domains are rated as weak.
Data synthesis
Our primary analysis consisted of a pooled estimate of prevalence across all studies using a random effects model.36 37 We also pooled data from studies separately under the model in order to calculate the pooled prevalence of FH in children (ages 0–19) and adults (>20 years of age). Where studies presented multiple diagnostic criteria, estimates derived from genetic testing were used in the analysis as this was thought to provide a more conservative estimate. Where studies derived estimates using DLCN criteria, we pooled reported cases of ‘definite’ or ‘probable’ FH to determine individual study estimates. Similarly, ‘definite’ or ‘possible’ FH diagnoses using Simon Broome criteria were pooled in the meta-analyses. Where multiple studies reported prevalence estimates from a single cohort, estimates were taken from the paper reporting the largest sample and the other paper excluded from the analysis. Potential influences on prevalence estimates were investigated using subgroup analyses and meta-regression. Where studies allowed, we descriptively compared prevalence estimates by age, sex, prevalence estimation method, study quality and geographical location within studies. We then assessed the influence of these factors on variation in the estimated prevalence using meta-regression models.
Statistical analysis
We calculated pooled prevalence figures with 95% CIs using the DerSimonian and Laird random effects model.37 In meta-analyses of prevalence using inverse variance methods, when the frequency estimate of a single study approaches the limits of prevalence (ie, 0% or 100% of the population), the variance for that study moves toward 0, leading to the resulting weight in the meta-analysis being overestimated.36 To accommodate for this, we conducted the meta-analysis with prevalence estimates that had been transformed using the double arcsine method.36 The final pooled result and 95% CIs were then back transformed and expressed as percentages for ease of interpretation. We assessed heterogeneity in our pooled analyses using the I2 statistic as it is not sensitive to the scale of effect size or the total number of studies included in the meta-analysis.38 Finally, publication bias was examined formally using Egger’s weighted regression, with significance set at p<0.10.39 Publication bias was also assessed visually using Begg’s funnel plot as well as a Doi plot.40 41 If publication bias was present, we used the trim and fill method to adjust for publication bias.40 Analyses were performed using the MetaXL add-in for Microsoft Excel (http://www.epigear.com). Forest plots were generated using DistillerSR Forest Plot Generator from Evidence Partners (https://www.evidencepartners.com/resources/forest-plot-generator/).
Meta-regression was used to discern the influence of age, sex, prevalence estimation method, study quality, geographical location, year of publication and study setting (ie, electronic health records versus general population registers) on our pooled prevalence estimate. We used Stata V.13.1 to perform the meta-regression analysis on the log scale of the back transformed effect size (ie, prevalence), with each trial weighting equal to that derived under the random effects model and between study variance estimated with the restricted maximum likelihood method. The log of the pooled prevalence estimate was used as the dependent variable whereas sample size, study quality scores, mean sample age and study proportions of female participants were used as continuous predictive variables. Categorical covariates such as prevalence estimation method and geographical location were dummy-coded and examined through a joint test for all dummy-coded covariates.
Sensitivity analyses
We conducted additional analyses to assess the robustness of our pooled prevalence estimate. We examined the impact of time on the diagnosis of FH by sequentially excluding studies published before the year 2000 and 2010. We also assessed the impact of study setting by comparing estimates derived from population-based databases with those in patient cohorts (ie, community clinics, patient registries, electronic health records). Finally, we excluded studies using LDL-C to diagnose FH as well as those from countries with known founder populations as both were likely to result in a higher pooled frequency.
Results
Study selection
Our search identified 4153 citations, of which 3574 were unique. After applying our inclusion and exclusion criteria, 90 articles progressed to screening at the full-text level, of which 21 articles were included in this review. The flow of included studies is presented in figure 1.

Flow of studies included in systematic review of heterozygous familial hypercholesterolaemia prevalence. FH, familial hypercholesterolaemia.
Characteristics of included studies
Twenty-one studies estimating point prevalence of FH were included in this review (table 1). The majority of these studies were European (n=9), while others were conducted in North America (n=4), Asia (n=2), Australia (n=3) and Africa (n=1). Two of the studies pooled data from international cohorts.10 42 Combined, they represented data from 28 countries across four continents. Studies representing multiple countries included data from coronary artery disease10 and dyslipidaemia cohorts.42 FH is overexpressed among those with coronary heart disease as well as statin-treated individuals.24 For these reasons, we elected against pulling country-specific data from these papers. Among all included studies, females comprised between 26.4% and 55.0% of the total sample. Four studies diagnosed FH using DLCN criteria,42–45 three studies used genetic sequencing,46–48 three studies used LDL-C measurements,49–51 one study used SBR criteria7 and one employed MEDPED criteria.6 Another four included studies reported prevalence estimates using more than one method for comparison.10 11 52 53 Prevalence estimates reported in individual studies ranged from 0.05% (95% CI 0.05% to 0.06%) to 5.62% (95% CI 5.44% to 5.79%). When evaluated by the EPHPP tool, most studies were rated as being moderate (n=7) or strong (n=13) in quality. On EPHPP domains, studies were most likely to receive weak ratings due to a low likelihood of representing the general population, a failure to account for missing participant data or adjust for relevant confounders (see online supplementary etable 7).
Characteristics of studies included in systematic review of FH prevalence
Meta-analysis
Overall pooled prevalence
Nineteen estimates were included in the meta-analysis of overall prevalence, representing 2 458 456 unique individuals.6 7 10 42–48 50 51 53–58 A further two studies reported data from cohorts represented by other studies within a shorter sampling frame, creating the potential for the overlap of cohorts.11 49 These estimates were excluded to avoid overweighting a population. The overall random effects pooled prevalence of FH was 0.40% (95% CI 0.29% to 0.52%) (figure 2).

Forest plot of overall pooled prevalence (%) of heterozygous familial hypercholesterolaemia. I2, between-study heterogeneity; LCL, lower confidence limit; POP, population; PREV, prevalence; UCL, upper confidence limit; WGHT, weight under the random-effects model. Note: prevalence estimates were derived using the double-arcsine method, back-transformed and expressed as percentages for ease of interpretation.
Prevalence of FH in adults
Sixteen prevalence estimates were included in the meta-analysis of adult prevalence, representing 2 431 053 unique individuals.6 7 10 42–48 53–57 The overall random effects pooled prevalence of FH was 0.40% (95% CI 0.29% to 0.54%) (see online supplementary etable 8).
Prevalence of FH in children
Combining four studies (n=27 403) which reported FH prevalence estimates in individuals aged under 19 (see online supplementary etable 9), we calculated a pooled prevalence of 0.36% (95% CI 0.28% to 0.45%), with little heterogeneity (I2=13.32%).43 50 51 58
Variation in prevalence by age
Six studies7 11 43 49 53 55 reported age-stratified data on the adult prevalence of FH, but only two of these presented data in forms amenable for pooled analysis (figure 3).7 53 All studies showed variation in FH frequency with age, with an increase in prevalence that peaked between ages 60 and 69 and declining thereafter, a trend reflected in our pooled estimates.

Age-stratified pooled familial hypercholesterolaemia (FH) prevalence estimates and 95% CIs. figure 3Error bars are representative of 95% CIs for each pooled estimate. Lower CIs are omitted; all cross 0%. I2, between-study heterogeneity; LCL, lower confidence limit; UCL, upper confidence limit.
Variation in prevalence by sex
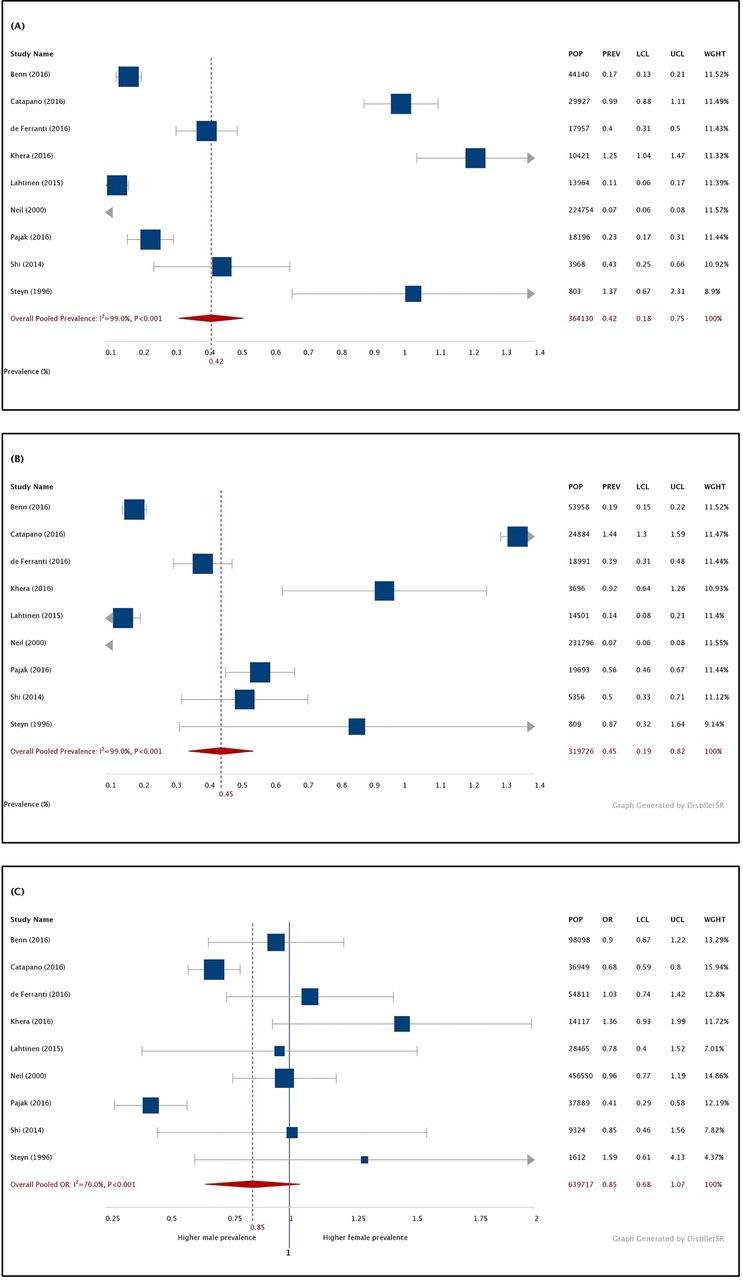
Nine studies presented prevalence figures by sex,7 10 42–44 46 47 52 53 most of which reported similar FH frequencies between men and women. Our pooled prevalence estimates (figure 4) were comparable between males (0.42%; 95% CI 0.18% to 0.75%; n=364 130) and females (0.45%; 95% CI 0.19% to 0.82%; n=319 726) (OR: 0.85; 95% CI 0.0.69 to 1.07; n=639 717).

(A) Forest plot of pooled prevalence (%) of heterozygous FH in the male population. (B) Forest plot of pooled prevalence (%) of FH in the female adult population. (C) Forest plot of pooled OR of male:female FH prevalence. FH, familial hypercholesterolaemia; I2, between-study heterogeneity; LCL, lower confidence limit; POP, population; PREV, prevalence; UCL, upper confidence limit; WGHT, weight under the random-effects model. Note: prevalence estimates were derived using the double-arcsine method, back-transformed and expressed as percentages for ease of interpretation.
Variation in prevalence by geographic location
When FH was analysed by continent (figure 5), European (seven studies; n=1 957 002) and Asian studies (one study; n=9324) tended to report lower prevalence estimates than our overall pooled prevalence estimate, while North American (three studies; n=236 537) and Australasian (two studies; n=175 512) studies reported estimates comparable to it. The one study from South Africa (n=1612) reported a greater pooled FH prevalence than our pooled estimate, as did studies of international cohorts.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Forest plot of overall pooled prevalence (%) of heterozygous familial hypercholesterolaemia stratified by population geography. I2, between-study heterogeneity; LCL, lower confidence limit; POP, population; PREV, prevalence; UCL, upper confidence limit; WGHT, weight under the random-effects model. Note: prevalence estimates were derived using the double-arcsine method, back-transformed and expressed as percentages for ease of interpretation.
Variation in prevalence by diagnostic criteria
Frequencies from studies in the DNA-based analysis subgroup were comparable to the pooled prevalence estimate (0.40%; 95% CI 0.24% to 0.58%) while DLCN (0.46%; 95% CI 0.25% to 0.70%) and LDL-C-based estimates (0.45%; 95% CI 0.34% to 0.57%) tended to report slightly higher frequencies (see online supplementary efigure 1). Of two studies exclusively using SBR7 or MEDPED6 criteria, both reported lower frequencies than our pooled prevalence estimate.
Variation in prevalence by study quality
When stratified by study quality ratings, studies rated strong had a lower estimate of FH prevalence with greater precision (0.33%; 95% CI 0.24% to 0.43%) than studies rated moderate in quality (0.75%; 95% CI 0.29% to 1.29%) or low quality (0.37%, 95% CI 0.12% to 0.74%) (see online supplementary efigure 2).
Meta-regression analyses
Considerable heterogeneity existed between studies (I2: 99.34%; 95% CI 99.24% to 99.44%). The results of eight meta-regression analyses (table 2) showed little evidence of an effect of age (p=0.79), sex (p=0.17), sample size (p=0.06), diagnostic criteria (p=0.23), study setting (p=0.50), quality (p=0.82) or year of publication (p=0.52) on our pooled prevalence estimate. Joint meta-regression tests showed significant differences in prevalence estimates among categories of studies when stratified by geographical location (p=0.04). Major asymmetry was present in both Begg’s funnel plot and the Doi plot (see online supplementary efigure 3) and the results of Egger’s test suggested that publication bias may have been present (p<0.001).59 When we used the trim and fill method to control for publication bias, nine additional studies were generated with estimates comparable to or lower than our pooled prevalence estimate, bringing the pooled prevalence of FH to 0.20% (95% CI 0.10% to 0.40%).
Meta-regression analyses for pooled estimate of familial hypercholesterolaemia prevalence
Sensitivity analyses
Pooled prevalence estimates were broadly consistent in seven sensitivity analyses (see online supplementary etable 10). Studies estimating FH prevalence in patient cohorts (0.33%; 95% CI 0.21% to 0.47%) tended to report lower frequencies than those in large population-based samples (0.45%; 95% CI 0.26% to 0.68%). Heterogeneity of these estimates was significant and comparable (>99%).
Discussion
Our meta-analysis of 19 cohort studies including 2 458 456 unique individuals found an FH prevalence of 0.40% in the general population. This suggests that as many as 1 in 250 individuals may be affected by FH (95% CI 1 in 345 to 1 in 192), equating to nearly 30 million people worldwide.60 This is a higher frequency than observed in prior reports and supports current thinking that FH is underdiagnosed, and thus likely undertreated in the general population.61 This is further supported by sensitivity analyses in which patient cohort studies were found to report lower prevalence estimates than those using large population databases.
Interestingly, we detected a slightly lower prevalence of FH in those aged 0–19 (1 in 278; 95% CI 1 in 345 to 1 in 222). Further, FH prevalence tended to increase with age. This trend runs counterintuitively to expectations given that FH is a genetic condition with a high risk of CVD-related mortality—frequency estimates should be comparable in adults and children save for age-related declines in prevalence associated with premature mortality. Our findings may be explained by insufficient dyslipidaemia screening in children and adolescents.62–64 Indeed, follow-up data from the Simon Broome FH registry, following more than 300 000 patients found that only a quarter of affected patients received diagnoses by middle age, with the highest rates of underdiagnosis among children and adolescents.7 However, LDL-C levels also rise with age, making it likely for older individuals to be diagnosed using established clinical criteria. It remains possible that the disparity in prevalence may be due to the inability of population-based studies to account for age-related increases in LDL-C and the reduced sensitivity this confers in detecting FH.65
Our finding that FH affects males and females equally has important implications. Many cases of FH are diagnosed following the first cardiac event, which has a later onset for women relative to men.27 This makes it possible that women with FH may go unrecognised for longer. Yet, more women may be expected to qualify for diagnosis using clinical characteristics at later ages, primarily due to the delayed onset of coronary artery disease. Whether delayed FH detection in women relative to men confers poorer clinical outcomes has yet to be formally explored in the literature. However, one of our included studies observed that after age 60, higher proportions of women met criteria for an FH diagnosis, suggesting that many men with FH had died at an earlier age.11 Identifying sex-related differences in FH presentation may allow for earlier FH diagnosis and represents an important clinical priority. New diagnostic criteria developed through improved use of routinely collected health data may make this possible.66
We also found lower prevalence reports in Europe relative to regions elsewhere. Thus far, much of the regional variation in FH prevalence has been attributed to the presence of founder populations. Founder effects occur when subpopulations are formed by the immigration of ‘founder subjects’, leading to a higher proportion of individuals who share a mutation in subsequent generations due to genetic drift.13 Though influenced by a predominance of European studies, our review suggests the potential for variations in FH frequency between countries extending beyond founder effects. This is important given that for many of the world’s countries, rates of FH still remain unknown. This includes North America, where studies from USA comprise the evidence base for ascertaining study prevalence. CVD remains the leading cause of death worldwide67 and, left untreated, nearly 85% of males and 50% of females with FH are expected to suffer coronary events prior to age 65.27 Thus, greater efforts should be made to explore region-specific frequencies of FH prevalence and more accurately characterise disease burden. Accurate prevalence estimates, augmented by recent big data approaches and the introduction of International Classification of Diseases, 10th Revision codes for FH should facilitate increased awareness and improved management.
How FH should be identified remains an area of continued debate. A number of organisations have recommended universal lipid screening in childhood as a strategy to identify FH.68–70 However, a recent report by the US Preventive Services Task Force concluded that there was ‘inadequate direct evidence on the benefit of screening for FH’.71 In addition, these programmes come with the added risks of potential overdiagnosis, fiscal and non-fiscal health system burden and adverse psychosocial impacts for children and families.71 As an alternative, some European countries have developed genetic FH screening strategies. However, such programmes are neither currently universally accessible nor deemed to be cost-effective.8 21–23 DNA-based identification may also fail to capture individuals with undiscovered mutations or those with polygenic forms of FH that still demonstrate the clinical phenotype.72 Finally, the diagnostic accuracy of these programmes has been challenged by findings that up to 30% of estimated cases may not be identified in countries with some of the most robust screening programmes, due to lack of index cases to inform cascade screening.73 In light of these limitations, the high degree of concordance between our pooled prevalence estimates derived through DLCN and DNA-based analyses are clinically important. Due to a simplified approach—facilitated by the use of readily observable clinic characteristics and biochemical parameters—DLCN criteria may facilitate the more ready identification of patients affected by FH in primary care. Though other clinical criteria may have comparable clinical utility, our study currently provides insufficient evidence in strong support of them. Regardless, improving the identification of FH and mitigating CVD and mortality requires a multifaceted approach involving clinical, biochemical and genetic parameters.
These findings provide new insights into FH prevalence. Yet, they should be interpreted in light of some important limitations. First, despite an extensive search strategy, we included only peer-reviewed English language studies indexed in six online databases and it remains possible that other relevant studies went unpublished or were indexed in other languages, in print repositories or within the grey literature.74 Second, we did not contact study authors for additional data or clarifications of their published studies. While this was counterbalanced in part by the use of a tool with high inter-rater agreement for quality assessment,75 agreement levels between reviewers and authors have yet to be explored with the EPHPP tool. Third, while geographical location of our included studies was significantly associated with variance in FH prevalence, our analyses possessed a considerable amount of between-study heterogeneity, the majority of which remains unexplained. This may be attributed to limited power in our meta-regression analyses due to small numbers of observations.38 In which case, our subgroup analyses provide more credible insight into the sociodemographic variation of FH prevalence though even these are limited by the lack of interaction tests in our subgroup analyses. It is important to note that the high degree of heterogeneity in our meta-analyses does not imply imprecision in our prevalence estimate.38 Indeed, a key strength of our study is its sample size and the greater power and precision it conferred to our analyses. The heterogeneity between studies are thus more likely reflective of real differences in study populations, designs and outcome measurements.36 This heterogeneity was anticipated and accommodated for through random effects meta-analysis.
Conclusion
Our systematic review found that FH currently affects 1 in 250 people in the adult population. While FH affects males and females equally, regional and age-specific variations exist in FH frequency. With the range of treatment options available for this condition increased, particularly with the recent advent of PCKS9 inhibitors, greater efforts should be made to identify individuals who could stand to benefit from therapy.
Acknowledgments
The authors would like to thank staff at the Institute for Clinical Evaluative Sciences for logistical support of this systematic review.
References
Footnotes
Contributors LEA conceived and designed the study, conducted the study, provided methodological support, conducted the analyses, interpreted the results and wrote, read and edited the manuscript. JG interpreted the results, read and edited the manuscript. SDS, RLR and JMA conducted the study and read and edited the manuscript. AC conceived and designed the study, provided methodological support and read and edited the manuscript. JVT conceived and designed the study, provided methodological support, interpreted the results, guided the analysis and read and edited the manuscript.
Funding LEA was supported by the Comprehensive Research Education for Medical Students Scholar Program at the University of Toronto. JG holds the McGill/Novartis Chair at McGill University. JVT is funded by a Canada Research Chair in Health Services Research from the Canadian Institutes of Health Research (CIHR) and an Eaton Scholar Award from the Department of Medicine at the University of Toronto. This research is also funded by an Institute of Circulatory and Respiratory Health-Canadian Institutes of Health Research Chronic Diseases Team grant (no. TCA 118349) to the Cardiovascular Health in Ambulatory Care Research Team (http://www.canheart.ca) and the Institute for Clinical Evaluative Sciences (ICES), which is funded by an annual grant from the Ontario Ministry of Health and Long-Term Care (MOHLTC). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Additional data are presented in supplemental files.