Article Text

Abstract
Introduction Pediatric sickle cell disease, highly prevalent in sub-Saharan Africa, carries great morbidity and mortality risk. Limited resources and monitoring make management of acute vaso-occlusive crises challenging. This study aims to evaluate the efficacy and safety of subdissociative intranasal ketamine as a cheap, readily available and easily administered adjunct to standard pain therapy. We hypothesise that subdissociative, intranasal ketamine may significantly augment current approaches to pain management in resource-limited settings in a safe and cost-effective manner.
Methods and analysis This is a multicentred, randomised, double-blind, placebo-controlled trial enrolling children 4–16 years of age with sickle cell disease and painful vaso-occlusive pain crises. Study sites include two sub-Saharan teaching and referral hospitals with acute intake areas. All patients receive standard analgesic therapy during evaluation. Patients randomised to the treatment arm receive 1 mg/kg intranasal ketamine at onset of therapy, while placebo arm participants receive volume-matched intranasal normal saline. All participants and clinical staff are blinded to the treatment allocation. Data will be analysed on an intention-to-treat basis. Primary endpoints are changes in self-report pain scales (Faces Pain Scale-Revised) at 30, 60 and 120 minutes and rates of adverse events. Secondary endpoints include hospital length of stay, total analgesia use and quality of life assessment 2–3 weeks postintervention.
Ethics and dissemination The research methods for this study have been approved by the Cameroon Baptist Convention Health Board Institutional Review Board (IRB2015-07), the Tanzanian National Institute for Medical Research (NIMR/HQ/R.8a/Vol. IX/2299), Muhimbili National Hospital IRB (MNH/IRB/I/2015/14) and the Tanzanian Food and Drugs Authority (TFDA0015/CTR/0015/9). Data reports will be provided to the Data and Safety Monitoring Board (DSMB) periodically throughout the study as well as all reports of adverse events. All protocol amendments will also be reviewed by the DSMB. Study results, regardless of direction or amplitude, will be submitted for publication in relevant peer-reviewed journals.
Trial registration ClinicalTrials.Gov, NCT02573714. Date of registration: 8 October 2015. Pre-results.
- sickle cell disease
- Vasoocclusive crisis
- Pain
- Ketamine
- Intranasal
- Opioids
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Strengths and limitations of this study
Multicentred for wider applicability and larger patient volume.
Participants and clinical staff blinded to intervention.
Utilises ketamine, an easily accessible and readily available intervention.
Lack of atomiser device for intranasal administration.
Both study sites are referral centres and may not represent typical population.
Unclear utility in resource-rich environments.
Utility of self-pain report scores.
Introduction
Sickle cell disease (SCD), the most common inherited haemoglobinopathy worldwide, affects approximately 70 million people globally with the vast majority in sub-Saharan Africa.1–3 Acute vaso-occlusive crisis (VOC) increases mortality and drives hospital admissions for persons with SCD.4–7 Across Africa, children with the haemoglobin SS form of SCD have a 50%–90% chance of early death, including in Nigeria, where the average life expectancy is approximately 21 years of age.8–11 Despite advances in maintenance therapy, painful vaso-occlusive episodes remain common, resulting in frequent presentation to acute care settings. A recent cross-sectional study from the Democratic Republic of the Congo shows that almost half of paediatric patients with SCD seek emergent medical care three or more times annually for VOC.12 Recurrent healthcare visits burden the local economy, and healthcare costs are anticipated to rise as the global burden of SCD increases.1–3 9–13 Treatment options for VOC are limited, relying heavily on non-steroidal anti-inflammatory drugs (NSAIDs) and opioids. Opioids pose a great risk of respiratory depression, the complications of which are magnified in resource-limited settings where access to monitoring and staffing are limited. Safe, cost-effective and readily available adjunctive therapies for VOC, which improve pain management while limiting resource utilisation, would be of great benefit in these settings.
Ketamine is cheap, safe, easily accessible, stable at room temperature and recommended by the WHO as an essential medication.14–19 Typical side effects at higher, dissociative dosing commonly used for moderate sedation include tachycardia, hypertension, nausea, nystagmus and emergence phenomena, defined as hallucinations or recovery agitation.18 20 Respiratory adverse events, such as apnoea and laryngospasm, are rare even at these higher doses and attributed to high-dose, rapid intravenous administration.20 Lower dose, subdissociative ketamine provides analgesia with minimal risk of depressing the respiratory system.18 21 22 Intravenous and intranasal (IN) ketamine have proven benefit for analgesia in prior trials of patients in the following settings: paediatric prehospital medicine, paediatric emergency medicine, chronic pain, paediatric oncology and battlefront.23–31 In 2010, Zempsky et al published the first case series on subdissociative ketamine infusions for paediatric SCD VOC.32 Subsequent studies corroborate that ketamine provides clinically significant analgesia for SCD pain and also reduces opiate requirements.17 32–36 Additional studies indicate that ketamine prevents opioid-induced hyperalgesia.37–40 The current study will examine the potential benefit of adjunctive subdissociative IN ketamine analgesic therapy in resource-limited settings.
There is strong precedent for using IN ketamine as a safe and efficacious analgesic.24 41 The PICHFORK (Pain In Children Fentanyl or Ketamine) Trial compared the analgesic effects of IN fentanyl (1.5 mcg/kg) to IN ketamine (1 mg/kg), concluding that IN ketamine provides equivalent analgesia to IN fentanyl with mild side effects of dizziness, mild drowsiness, nausea, dysphoria, itchy nose and bad taste in the mouth.41 Twenty-eight patients in the ketamine treatment arm experienced adverse events, as compared with 15 in the fentanyl arm, but all were minor events, and none posed safety concerns or required additional interventions.41 Based on pharmacodynamic studies of ketamine and the precedent set from previous trials, IN ketamine dosed at 1 mg/kg produces a bioavailable dose of 0.4–0.8 mg/kg, which is safely within the accepted subdissociative dose range.24 37–45 This side effect profile and bioavailability allow for safe and reliable dosing in resource-limited settings.
Subdissociative ketamine may therefore be an ideal medication for vaso-occlusive pain management in sub-Saharan Africa. IN administration is rapid and cheap. IN administration reduces time to medication delivery, avoids painful intravenous line placement or intramuscular injection and reduces use of staffing resources.46 Mucosal atomisers enhance efficacy and tolerance, but they are cost-prohibitive in resource poor settings.47–50 However, there is precedent for using IN analgesia without atomisers, with similar efficacy.51 As such, we hypothesise that IN ketamine will be a safe and effective adjunct to standard VOC pain management for children with SCD treated in resource-limited settings.
We propose a multicentred, randomised, double-blind, placebo-controlled trial evaluating the efficacy and safety of adjunctive IN ketamine as compared with standard VOC pain management. The results will add to the limited published data on ketamine in the management of paediatric SCD vaso-occlusive pain crises, use of IN ketamine for acute paediatric pain and novel, cost-effective SCD pain management strategies in resource-limited settings.
Methods and analysis
Aims and hypotheses
Primary hypothesis
Subdissociative IN ketamine in combination with standard treatment provides safe and rapid pain relief as compared with standard treatment alone among paediatric sickle cell patients age 4–16 years with pain crises in resource-limited settings.
Primary efficacy aim
Compare the efficacy of standard pain management plus adjunctive subdissociative IN ketamine versus standard pain management, as measured by self-report Faces Pain Scale-Revised (FPS-R) at time points during the first 2 hours postintervention.
Safety aim
Compare the frequencies of side effects, adverse events, and serious adverse events over 2 hours among paediatric SCD patients with VOC randomised to receive standard pain management or standard management plus adjunctive subdissociative IN ketamine.
Secondary efficacy aims
Compare hospital length of stay, total analgesia use (morphine equivalents for opioids and total NSAID or paracetamol (PCM) use/kg body weight) and quality of life assessments (as measured by the Pediatric Quality of Life Inventory-Sickle Cell Disease Module (PedsQL-SCD)) at 2–3 weeks postintervention among paediatric SCD patients with VOC assigned to standard therapy plus adjunctive subdissociative IN ketamine versus standard therapy alone.52
Trial design
This is a multicentred, randomised, double-blind, placebo-controlled drug trial evaluating the efficacy and safety of standard VOC pain management plus adjunctive subdissociative IN ketamine as compared with standard VOC pain management in paediatric SCD patients with VOC in resource-limited settings. This study protocol is designed and written in compliance with the SPIRIT 2013 Checklist to promote quality study conduct and transparent interpretation and reporting of study results.
Study setting
The study will be conducted at two tertiary care, referral centres in sub-Saharan Africa. The first site is the emergency department (ED) and adjacent inpatient paediatric ward of Muhimbili National Hospital (MNH) in Dar es Salaam, Tanzania. MNH is the largest tertiary care centre in Tanzania, receives 80–140 critically ill patients daily, cares for greater than 400 sickle cell patients monthly and hosts the only emergency medicine residency training programme in the country. The second study site is Mbingo Baptist Hospital (MBH) paediatric outpatient clinic/urgent care and adjoining inpatient paediatric ward in Mbingo, Northwest Province, Republic of Cameroon. MBH is the primary teaching and referral centre for the Cameroon Baptist Convention Health Board hospital network, hosts internal medicine and surgical residencies and receives greater than 200 patients daily in the acute intake areas. MBH provides care for greater than 200 paediatric patients with sickle cell annually. Both facilities have 24 hours per day coverage with residents and attending physician supervision.
Eligibility criteria
Paediatric patients age 4–16 years with SCD presenting with VOC requiring analgesia.
Exclusion criteria
Patients with the following characteristics will be excluded from study enrolment:
anatomic variations of nose precluding IN medication administration
ketamine allergy
non-verbal or obtunded
pregnant
schizophrenia
any patient with presumed complications of SCD beyond pain crisis:
acute chest syndrome
sepsis
stroke
splenic sequestration
pulmonary embolism
acute osteomyelitis
lack of family/personal phone to facilitate follow-up interview
Recruitment and consent
Patient screening and enrolment begins at acute intake area triage and is overseen by the site principal investigator and performed by trained study personnel. A screening log will document all eligible patients screened and approached along with the reasons for any exclusions. Standardised consent forms were developed and translated by bilingual study staff into the languages most common to the encountered study population, including English, French and Swahili. Fluent study personnel review the consent forms in the participant’s native tongue and a standard study intake form (case report form) is used to collect demographic data, vital signs and medication administration records throughout the encounter. Assent is obtained for patients 8 years and older as approved by both site IRBs.
Interventions
Patients randomised to the intervention arm will receive 1 mg/kg subdissociative IN ketamine per the standard dosing table, marking time zero (supplementary appendix A). Those randomised to the placebo arm will receive volume matched IN 0.9% normal saline per the standard dosing table, marking time zero (supplementary appendix A). A 1 mL syringe, without needle, is inserted gently into the nares with the patient sitting upright. Volumes of ≤0.75 mL will be administered in a single nare, while volumes >0.75 mL will be divided between both nares. Patients who are unable to inhale the medication nasally will receive drip administration of the same volume while recumbent on the bed. Consenting/assenting participants who refuse nasally administered medications despite appropriate consent/assent will be removed from study protocol, although their data will be included in the intention-to-treat analysis.
Study medications
Ketamine (50 mg/mL) and 0.9% normal saline are on the formulary at both study hospitals and are stored in accordance with hospital protocol and procedures.
Cameroon
Ketamine—ketamine chlorhydrate injection USP (50 mg/mL); manufacturer: Popular Pharmaceuticals, Dhaka, Bangladesh.
Normal saline—0.9% sodium chloride; manufacturer: Shijiazhuang No. 4. Pharmaceutical, Hebei, China.
Tanzania
Ketamine—ketamine (50 mg/mL); manufacturer: RotexMedica, Germany.
Normal saline—0.9% sodium chloride; manufacturer: Abacus Parenteral Drugs, Kampala, Uganda.
Concomitant medications
All patients will receive the assigned IN medication and oral ibuprofen (10 mg/kg per os (PO) × one dose, max dose: 600 mg), marking time zero. If an NSAID has been taken within 4 hours of hospital encounter, oral PCM (15 mg/kg PO × 1, max dose: 1000 mg) is administered along with IN medication at time zero. Additional analgesic interventions are administered as per standard VOC pain management at the sole discretion of the treating physician (supplementary appendix B), and all subsequent interventions are recorded on the participant’s case report form.
Measures and outcome measures
The primary efficacy outcome measure will be differences in FPS-R pain scale scores at 30 min, 60 min and 120 min from administration (time zero) as compared between the intervention and placebo groups (supplementary appendix C). FPS-R scores will be measured at 0, 30, 60 and 120 min, or until time of discharge home, whichever is shorter. The primary safety aim will be the frequency of side effects and adverse events as queried via a standard checklist at time points 30, 60 and 120 min, or as clinically indicated (appendices D and E). Patients are monitored for adverse events for 2 hours after IN medication administration, or until discharge home, whichever is first.
The secondary outcome measure of hospital length of stay will be defined from the time of the study drug administration to the time of discharge. The secondary outcome measure of total analgesia use will be defined as morphine milligram equivalents and NSAID/PCM dose per kg body weight. The secondary efficacy outcome measure of quality of life, as measured by PedsQL-SCD scores, will be performed by phone interview with trained study clinicians 14–21 days postintervention compared between the intervention and placebo groups (supplementary appendix F).52 Sections in the original PedsQL-SCD pertaining to ‘worrying,’ ‘emotions’ and ‘communication’ were withdrawn from the study protocol based on cultural applicability and feasibility.52 See figure 1 for a flow diagram of the study protocol from enrolment through discharge and phone call follow-up.
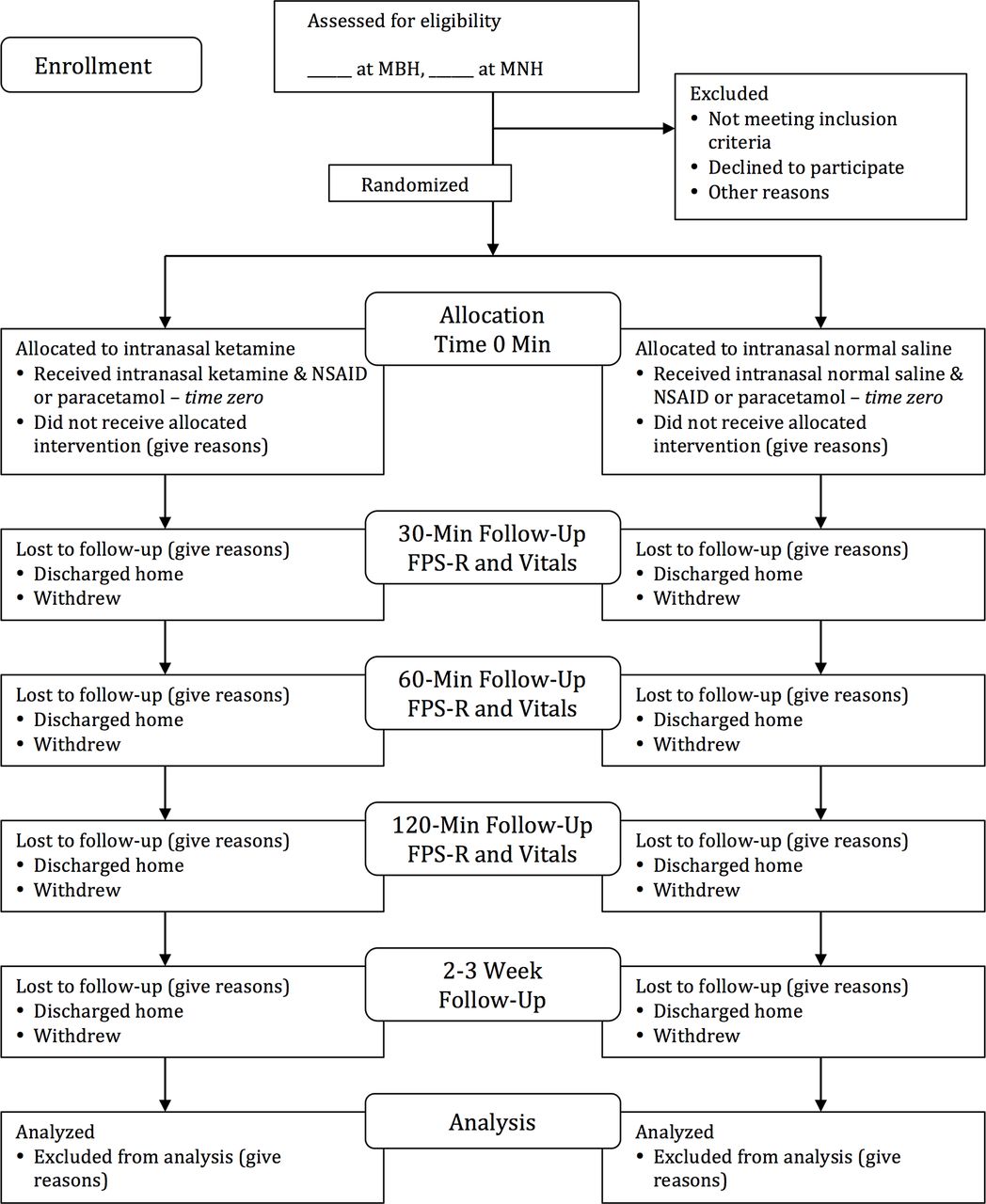
{kind=link}
Study flow diagram.
Sample size
Power analysis is based on two-sample t-test to compare the mean pain intensity scores in paediatric patients receiving two different treatments for sickle cell pain management. The goal of the analysis is to provide a rough estimate of the sample sizes needed to detect varying changes in pain scores given the mean and SD of a similar study.
A 2008 publication by Palermo et al (‘Daily Functioning and Quality of Life in Children with Sickle Cell Disease Pain: Relationship with Family and Neighborhood Socioeconomic Distress’) was used as the reference for the mean pain score (4.5) and SD (1.9).53 With n=77 per group, we will have 90% power to detect a difference of 1 on the FPS-R at 30 min, assuming SD=1.9 and two-sided alpha=0.05. We do not anticipate missing data due to the short duration of follow-up for each child; therefore, we have not inflated the sample size for attrition.
Blinding, allocation and concealment
Study packets containing identical prefilled syringes of either ketamine (intervention) or normal saline (placebo) are prepackaged in batch ahead of time by unblinded pharmacy personnel in accordance with the permuted block randomisation provided by the study statistician. The study statistician will generate randomisation lists using permuted block randomisation stratified by location. The lists will be provided to the study pharmacists for dispensing and assigning in the order specified with unique numbers, which will be recorded on each child’s case report form. The randomisation key that links names to study IDs will be securely stored in a study lock box within the research-specified office space at each centre. Only the study pharmacist who performs the randomisation will have on-site access to this list. Study packets are maintained according to local pharmacy standards and administered by study personnel on enrolment. Only the unique study ID on the packet and packet contents links the identification number to the contents of the enclosed syringes. Both ketamine and normal saline are colourless, salty-tasting liquids. Syringes and syringe contents are identical in appearance and volume regardless of study group assignment, maintaining blinding of administering clinician and participant. Additional components in the study packet include the standard dosing table for IN drug administration, standardised SCD treatment algorithm and participant case report form (appendices A and B). On enrolment, trained study staff will obtain the next sequential enrolment packet, labelled with study protocol number and containing the aforementioned contents, from the pharmacy located in proximity to the treatment areas and staffed by dedicated pharmacy personnel. Packets will be replaced every 3 months in accordance with the shelf life for ketamine and institutional policy. All unblinded study documents, including drug logs and randomisation schedule, are securely stored and maintained by the on-site research study pharmacists.
Statistical methods
All analyses will be conducted using intention to treat. For the FPS-R, we will compare the two treatment arms using a linear mixed model with fixed effect for group, time and group*time interaction with random effect for the subject to adjust for correlation among measures from the same child. We will use a contrast to test the two groups at 30 min and at subsequent time points. We are assuming the FPS-R measures are normally distributed. If they are not, we will transform them using log transformation, square root or inverse, depending on the skewedness of the data. For side effects and adverse events, we will report and compare the proportion of children experiencing the event after administration between the two groups using χ2 or Fisher’s exact tests. Length of stay and amount of other pain medications will be compared using two sample t-tests or Wilcoxon rank sum tests (if highly skewed). We will compare the four incorporated dimension scales of PedsQL-SCD between the two groups using a two-sample t-test or Wilcoxon rank sum depending on the distribution of the subscale. Following the recommendation of the PedsQL-SCD scoring, if more than 50% of items in a scale are missing, the scale scores will not be computed. If 50% or more of the items are completed, we will impute the mean of the completed items in a scale.54
Monitoring
The study Data and Safety Monitoring Board (DSMB) will operate in accordance with the guidelines established by the United States Food and Drug Administration in ‘Guidance for Clinical Trial Sponsors: Establishment and Operation of Clinical Trial Data Monitoring Committee', jointly published by the Center for Biologics Evaluation and Research, Center for Drug Evaluation and Research and Center for Devices and Radiological Health for the FDA, Office of Management and Budget Control No. 0910–0581, March 2006, expiration date 31 October, 2015 (updated guidance will be used as available).55 The study is unfunded, and the members of the DSMB have no competing interests in the project. DSMB member details are provided in supplementary appendix G. The DSMB will review study implementation and the occurrence of adverse events. Reports of all serious adverse events will be forwarded to the DSMB for emergent review (supplementary appendix E). Serious adverse events trigger unblinding of the affected participant to prevent future exposure to the implicated agent, although study treatment will not be altered. Should the DSMB identify any clinically important safety concerns, it will make recommendations related to these findings to the investigators and IRB, including (1) continue without modifications, (2) continue with modifications or (3) terminate the study. Any associated protocol modifications will be communicated via email within 48 hours to site investigators and associated site regulatory bodies and then confirmed by email or phone communication.
Ethics and dissemination
Ethics approval and consent to participate
Permission was obtained from both the International Association for the Study of Pain and JW Varni (PROQOLID, Mapi Research Trust) for use of the FPS-R and the PedsQL-SCD, respectively. All protocol amendments will be verified by the DSMB and reviewed with clinical staff at each site within 7 days of approval, as needed. Local IRB approval will be obtained annually and on an as-needed basis for potential protocol changes.
Ethical approval was obtained from the Cameroon Baptist Convention IRB in October 2015 (IRB2015-07). Subsequent approval was obtained from the Tanzanian National Institute for Medical Research (NIMR/HQ/R.8a/Vol. IX/2299) in September 2016, MNH (MNH/IRB/I/2015/14) in November 2016 and the Tanzanian Food and Drugs Authority (TFDA0015/CTR/0015/9) in November 2016.
Consent for publication
Not applicable.
Availability of data and material
All participant information is collected and maintained in a confidential manner. Case report forms are deidentified and labelled with only a study protocol number. Study data will be collected and managed using Research Electronic Data Capture (REDCap) tools hosted at MBH and MNH.56 REDCap is a secure, web-based application designed to support data capture for research studies, providing (1) an intuitive interface for validated data entry; (2) audit trails for tracking data manipulation and export procedures; (3) automated export procedures for seamless data downloads to common statistical packages; and (4) procedures for importing data from external sources. Trained personnel at each study site will directly enter deidentified data from the case report forms into the REDCap database. Site investigators and REDCap specialists oversee data transfer to ensure accuracy and quality of the data. During and after the study, all data forms will be kept in locked cabinets or on encrypted computers and servers at each site, accessible only by the site principal investigator and study team.
Dissemination
Dissemination of research data will be provided at both regional hospitals and subsequently through scientific conference(s) and publishable manuscript(s) regardless of magnitude or direction of the observed results.
Discussion
This study relied on existing research infrastructure within both African hospitals. In Tanzania, Carolinas Medical Center Department of Emergency Medicine supports a research collaboration including physician investigators and study coordinators. Investigators from Carolinas Medical Center have a history of successful enrolment of MNH ED patients in several prior and ongoing investigations.57–61 Our institution also has direct ties to MBH in Cameroon, where one of our study authors previously served as the chief of paediatrics. Current and former personnel from MBH have successfully collaborated on numerous national and international research investigations.62–67 The pre-existing infrastructure present at both sites, combined with personal knowledge and established relations between study sites, allows for study feasibility and anticipated success.
Trial status
Patient enrolment began in December 2015 in Cameroon after the Cameroon Baptist Convention IRB initially reviewed and approved the study protocol (supplementary appendix H). Due to hospital personnel changes, enrolment was temporarily held but resumed in July 2016 and is ongoing. In Tanzania, the study has been reviewed and approved by MNH IRB, the National Institute for Medical Research and the Tanzanian Food and Drugs Authority (appendices I, J and K). Enrolment began in March 2017. To date, 19 patients have been enrolled. We anticipate completion of enrolment by December 2017.
Acknowledgments
Special thanks to Sui Manasius (Head of Pharmacy, MBH), Samuel Ngum (Assistant Administrator, MBH), Elizabeth, Emmanuel and Ibrahim (research assistants, MBH), as well as Ms. Mtoke Uledi (in charge of Pharmacy at MNH ED), Dr. Faraja Chiwanga (Director of Research Training and Consultancy, MNH) and Mr. Helgod Sizya and Amani Kalebo (research assistants, MNH ED).
References
Footnotes
Contributors JRY, MSR and SR coconceived of the study. JRY, MSR, SR and CGM initiated study protocol design, and EN, EH, HRS and JAM served as implementation coordinators at their respective study sites, focused on identification, recruitment and enrollment, data collection and entry, and adherence to study protocol. CGM provided statistical expertise in both study design and data analysis. EN and HRS served as logistics experts within their site locations, liaised with pharmacy staffing and administration and helped to organise study-specific training for clinical staff in conjunction with JRY, the study PI. All authors contributed to development of the final manuscript and have approved its contents.
Funding This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors. Departmental funds from Carolinas Medical Center, Department of Emergency Medicine, are used for research assistant training and employment during the study period. Study medications and supplies are obtained through routine vendors as per hospital practice.
Competing interests No authors disclosed a relevant conflict of interest.
Patient consent Standardised consent and assent forms were signed by guardians and participants enrolled in the ongoing study. All data obtained from the study is deidentified prior to review and data analysis. This proposal, however, is a protocol paper explaining the process of implementing the study. No individual patient data is represented in this submission.
Ethics approval CBCHB IRB, MNH IRB, Tanzania FDA, and Tanzania NIMR.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement This is a protocol paper, and no analysed results are available at this time. In addition to anticipated forthcoming publication results, additional unpublished data will be available to interested parties via direct email communication with the lead investigator.