Article Text

Abstract
Introduction Irritable bowel syndrome (IBS) is characterised by the association of abdominal chronic pain with bowel habit disorders in the absence of identifiable organic disease. This is the first reason for consultation in gastroenterology, with an estimated prevalence of 10%–15% in industrialised countries. Although this is a benign gastrointestinal disease, its chronicity profoundly impacts the patient’s quality of life and causes considerable health spending. Actual medical treatments are poorly efficient on IBS-related abdominal pain, making it a major public health concern. The mechanisms causing IBS symptoms are unknown. Recent studies have shown the involvement of T-type channel in abdominal pain. We aim to evaluate the therapeutic potential of ethosuximide, a T-type channel blocker, on the abdominal pain of patients presenting an IBS.
Methods and analysis The IBSET trial is a randomised, controlled, parallel, double-blind and multicentre study. It is the first clinical trial evaluating the efficacy and safety of ethosuximide on abdominal pain related to IBS. Adults with IBS that report significant abdominal pain (≥4/10) at least for 3 months will be included. 290 patients will be randomly assigned to receive either ethosuximide or placebo over 12 weeks after 1 week of run-in period. The primary endpoint is the rate of responders (pain reduction ≥30% and Subject Global Assessment of Relief score ≥4). The intensity of abdominal pain will be assessed by an 11-point Numerical Rating Scale before and after 12 weeks of treatment and the score of the Subject Global Assessment of Relief scale at the end of treatment. The secondary endpoints are the safety of ethosuximide, the intensity and features of IBS and quality of life.
Ethics and dissemination The study was approved by an independent medical ethics committee (CPP Sud-Est VI, Clermont-Ferrand, France). The results will be published in a peer-review journal and presented at international congresses.
Trial registration number NCT02973542; Pre-results.
- ibs
- t-type calcium channel
- ethosuximide
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Strengths and limitations of this study
First clinical study assessing ethosuximide on abdominal pain related to IBS.
Randomised, parallel, double-blind and placebo-controlled study.
Larger study (290 patients).
Multicentre study.
Methodology consistent with the recommendations of the European Medicines Agency.
Eligible population close to clinical practices.
Intention-to-treat analysis.
Diabetic population not considered.
Absence of colorectal distension test.
Absence of Cav3.2 colonic mucosal expression assessment.
Short follow-up period.
Introduction
Irritable bowel syndrome (IBS) is characterised by abdominal pain, bloating and changes in bowel habit in the absence of an identifiable organic disease.1 2 This functional disorder is very common as its prevalence is estimated around 10%–15% in Europe and the USA.3–6 IBS chronicity severely impacts patients’ quality of life and generates large healthcare costs.7 8 The high prevalence and few effective treatments make this disease a public health concern. The mechanisms involved in visceral pain and IBS symptoms are still unknown. Recent studies indicate that alterations of the intestinal epithelial barrier is a key pathophysiological event in IBS, allowing maintenance of a low-grade mucosal inflammatory state, leading to visceral hypersensitivity (VH) and pain.9 10 This VH, highlighted by the colorectal distension test,11 is found in more than half of IBS patients.12–14 Hypersensitivity implies sensitisation of somatic or visceral primary afferent fibres, involving an increase in excitability. In several animal models of chronic pain, this hyperexcitability has been linked to a remodelling of ion channel expression patterns.15 16 Several voltage-gated ion channels are involved in neuronal firing, including T-type calcium channels, especially Cav3.2 member.17 18 We recently found in a case–control study that this Cav3.2 channel was overexpressed in the colonic mucosa of patients with IBS compared with asymptomatic controls.19 Moreover, it has been shown that the blockade of Cav3.2 channels, in an animal model mimicking IBS, prevented the development of colonic hypersensitivity.20 We aimed to evaluate the efficacy and the safety of a T-type calcium channel blocker—ethosuximide—in the treatment of abdominal pain in patients with IBS.
T-type calcium channels, ethosuximide and IBS
For 10 years, Cav3.2 channels have been extensively studied and shown as involved in nociceptive pain.18 21 Marger et al recently demonstrated, in an animal model of butyrate-induced VH, that Cav3.2 channels were involved in chronic visceral pain.20 They have shown that Cav3.2 channels were overexpressed in the dorsal root ganglion neurons innervating the colon and that their pharmacological blockade by ethosuximide prevented the development of colonic hypersensitivity. Recently, we demonstrated in a case—–control clinical study that Cav3.2 channels were overexpressed in the colonic mucosa of patients with IBS compared with asymptomatic controls.19 These channels are located particularly in peripheral nerve fibres, supporting their involvement in the development of colonic hypersensitivity. Cav3.2 channels appear to be an interesting pharmacological target to treat visceral pain related to IBS. The therapeutic effect of ethosuximide was studied in several animal models of chronic pain: traumatic neuropathy22–24 and chemotherapy-induced peripheral neuropathy,25 26 inflammatory27–30 and acute pain.29–32 Moreover, ethosuximide was shown to be a modulator of pronociceptifs effects of hydrogen sulfide,28 33 34 a gazotransmitter involved in VH related to IBS35 36 and a Cav3.2 channel activator.37 38 Thus, according to the results of these studies, ethosuximide, by its action on T-type calcium channels, seems to be a promising and innovative therapeutic in the treatment of abdominal pain in IBS patients.
Safety of ethosuximide in humans
According to the summary of product characteristics (SmPC), the safety profile of ethosuximide is similar with other antiepileptic drugs (AEDs). Frequently listed side effects are dyspepsia (nausea, epigastric pain, bloating and loss of appetite), dizziness, headache, ataxia, skin rash and vomiting. In adults, the effective dosage is 20 mg/kg/day, that is, 30 mL of syrup (1500 mg).
Summary of hypotheses and objectives
T-type calcium channels blockers have a major interest for the development of new symptomatic treatment of pain related to IBS. There is an opportunity to conduct an innovative proof-of-concept trial in patients with IBS with such a blocker, ethosuximide, which is currently available on the European market (to treat epilepsy). The demonstration of clinical efficacy would enrich our pharmacopoeia to treat pain related to IBS.
Methods and analysis
The present study is a randomised, controlled, parallel, double-blind and multicentre phase II pilot trial that evaluate the efficacy and safety of ethosuximide in patients with abdominal pain related to IBS. Two hundred and eighty-six patients are planned to be included among 10 clinical sites in France. The duration of the study is 16 weeks, including 1 week run-in period, 12 weeks of treatment and 3 weeks of follow-up.
Study objectives
The primary objective of this study is to evaluate the efficacy of ethosuximide versus placebo, administered in addition to background therapy, on intensity of abdominal pain related to IBS and to evaluate the score of the Subject Global Assessment of Relief (SGA scale).
The secondary objectives concern the effects of ethosuximide on:
the severity of IBS symptoms
the quality of life (physical and mental)
the safety of ethosuximide.
The study discontinuation rate and analgesics and transit regulator consumption will be assessed throughout the study.
Inclusion and exclusion criteria
Participants are adult patients with abdominal pain related to IBS diagnosed for more than 3 months and not relieved by the usual treatments (see details in box 1).
Inclusion and exclusion criteria of the study
Inclusion criteria
Man or woman aged 18 years or more.
Negative pregnancy test and effective contraception.
IBS defined by ROME IV criteria.
Treatment failure (11-point NRS abdominal pain≥4) for at least 3 months despite stable treatment for a month.
Patients affiliated to the regime of the French Social Security.
Patients able to deliver a free and informed consent.
Exclusion criteria
Breastfeeding.
Diabetic patients.
Chronic pain of greater intensity than the pain related to IBS.
Renal or hepatic impairment defined by significant liver function (liver enzymes >3 × normal values, cholestasis) and renal (modification of the diet in renal disease (MDRD) <60 mL/min) abnormalities.
Current or history of severe depression (hospitalisation, long-term antidepressant treatment).
Addiction to alcohol and/or drugs.
Patient already treated with antiepileptic.
Epilepsy.
Allergy to succinimide (ethosuximide, methsuximide, phensuximide).
Psychotic disorders.
Insufficient cooperation and understanding to adhere strictly to the conditions provided by the study.
Patients undergoing a measure of legal protection.
No therapeutic change will be generated by the protocol; patients will be treated with ethosuximide or placebo in addition to their current treatment of IBS. However, no therapeutic change for IBS will be allowed during the entire study. Only weak analgesics will be allowed (eg, paracetamol and non-steroidal anti-inflammatory drugs). Use of concomitant medications will be indicated by the patient on his logbook and during scheduled visits to the investigator. If the patient uses unauthorised treatment or changes his background treatment without prior authorisation, the patient will be excluded from the analysis because of a major deviation from the protocol.
Patients can be withdrawn of the study for any of the following reasons: modification of the analgesic therapy, intolerance to ethosuximide, withdrawal of consent, breach of protocol and significant adverse events.
Investigational medicinal product
Zarontin (Pfizer, ethosuximide)
The active substance is ethosuximide. This is an antiepileptic and a T-type calcium channel blocker. It aims to treat epileptic seizures or at least decrease their frequency or intensity. It is active on absence seizures and used alone or in combination with another AED in the treatment of generalised epilepsies. It is recommended by the SmPC to start with 10 mL (500 mg) of syrup from the age of 6 years. Depending on the response, the dosage should be increased very gradually by 5 mL (250 mg) of syrup every 4–7 days until complete control of the seizures. In adults, the effective dosage is 20 mg/kg/day, that is, 30 mL of syrup (1500 mg). The maximum dosage is 2000 mg per day (40 mL of syrup). In children, the effective dosage varies between 20 and 30 mg/kg/day, and the maximum dosage is 1000 mg per day.
In this study, ethosuximide will be administered daily during the dinner for 12 weeks. The dosage will be increased very gradually every 7 days, during 28 days, until reaching the maximum dose of 10 mL (500 mg) per day (from 2.5 mL to 10 mL/day, for details see figure 1). The choice of this dosage is based on a study currently conducted by our team (EDONOT study: NCT02100046) showing that ethosuximide doses ≥750 mg/day (15 mL) are poorly tolerated, whereas the 500 mg/day (10 mL) exhibits a clinically relevant analgesic effect associated with good tolerance in patients with neuropathic pain.

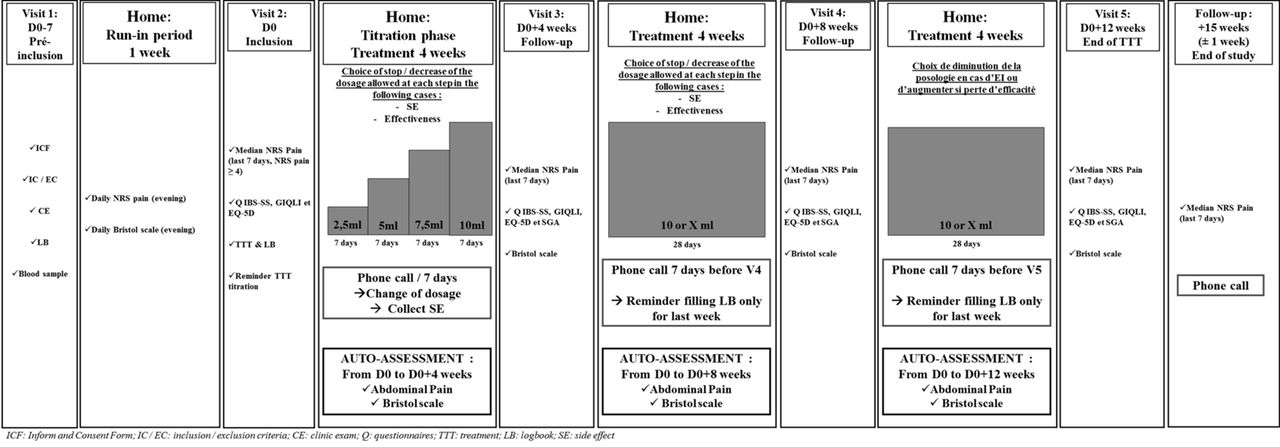{kind=link}
Study design overview. CE, clinic exam; ICF, inform and consent form; IC/EC, inclusion/exclusion criteria; LB, logbook; Q, questionnaires; SE, side effect; TTT, treatment.
However, if during the titration phase the patient has side effects related to ethosuximide, the investigator has the option to continue treatment at the lower dose level, if well tolerated, and this up to the end of the study. Moreover, stopping dosage escalation will be allowed in cases of effectiveness of the treatment. This pragmatic attitude corresponds to the current clinical practice and should reduce the risk of study discontinuation due to adverse events. It will also determine, in this indication, the optimal therapeutic dose.
Placebo
Placebo will be raspberry syrup containing the same sugar concentration and the same aroma as the Zarontin. The placebo will be taken for 12 weeks with the same administration modalities of the ethosuximide group.
Double-blind conditions
The staff of the investigation centre will not be allowed to open the boxes containing the therapeutic units. Only pharmacists from each centre will be able to deliver and recover therapeutic units for each visit. The therapeutic units will be packaged in similar bottles and in identical opaque boxes in order to preserve the double blind.
Study endpoints
Study endpoints were selected on the recommendation of the European Medicines Agency (International Council for Harmonisation guideline IBS: EMA/CHMP/60337/2013-CPMP/EWP/785/97 Rev. 1: for short treatment on all types of IBS).
Primary endpoint
Rate of responders to treatment: a patient will be considered as a responder if he satisfies two conditions: (1) a decrease of at least 30% of the score of abdominal pain (11-point NRS) compared with the score before treatment, and (2) a significant global improvement defined by a score of 4 or 5 on the SGA scale.
Abdominal pain intensity (11-point NRS)
This scale allows the patient to rate his abdominal pain from 0 to 10, with 0 is no pain and 10 the worst possible abdominal pain. The intensity of abdominal pain (the past 24 hours) will be measured daily (the evening) by the patient on the logbook, and the values will averaged for the 7 days preceding the two time points D0 (baseline) and D0 +12 weeks (last visit). The primary endpoint is the calculated difference Δ(%) = ((NRS(D0) – NRS(D0 +12 weeks)/NRS(D0)) × 100.
SGA Scale
The SGA Scale39 aims to assess the effectiveness of a specific treatment on symptoms of IBS. This scale consists of five levels of description answering the question ‘Please consider how you felt during the past treatment period in regard to your IBS, in particular your overall well-being, and symptoms of abdominal pain/discomfort and altered bowel habit’. Levels of description are declined such as: (1) worse, (2) not at all relieved, (3) somewhat relieved, (4) considerably relieved and (5) completely relieved.
These simple responses sign both satisfaction and dissatisfaction. This evaluation will be conducted during the last visit of the study (D0 +12 weeks).
Secondary endpoints
Health-related quality of life (HRQoL) (Gastrointestinal Quality of Life Index (GIQLI) and EuroQol five dimensions questionnaires (EQ-5D-3L))
HRQoL will be evaluated by the GIQLI and EQ-5D-3L (EuroQol group), which assess the physical and mental health of IBS patient.
The GIQLI questionnaire40 is a specific self-questionnaire for measuring the quality of life of patients with gastrointestinal disease. The questionnaire includes 36 items asking about symptoms, physical status, emotions, social dysfunction and effects of medical treatment.
The quality of life questionnaire ‘EQ-5D’41 is a non-specific questionnaire used to assess the health status and the impact of a disease on patient’s quality of life. The EQ-5D score is measured on five dimensions (mobility, personal autonomy, routine activities, pain and anxiety/depression) with three levels per dimension and a scale from 0 (worst imaginable state of health) to 100 (best imaginable state of health). These assessments are done four times at visit 2, 3, 4 and 5.
IBS severity (Irritable Bowel Syndrome - Severity Scoring System (IBS-SSS) and Bristol Scale)
IBS-SSS questionnaire42 is a self-administered questionnaire used to assess the severity of the specific symptoms related to IBS. This questionnaire consists of seven questions giving a score between 0 and 500 points. IBS intensity is considered mild for a Francis score less than 175, moderate for a score from 175 to 300 and severe if the score is above 300.
The scale of Bristol is a medical diagnostic tool created to classify the shape and consistency of human faeces. The scale has seven distinct categories. This assessment is done at home by the patient alone, every week before planned visits, on the daily logbook (the evening (before bed): stools number and consistency).
Genetic analysis of cytochromes P450
A blood sampling will be performed for the genetic analysis of the genes coding for CYP3A4 and CYP2E1, involved in the metabolism of ethosuximide.43 This genetic analysis will make it possible to demonstrate the existence (or not) of slow/fast metabolisers in our studied population.44 45 The demonstration of slow/fast metabolisers will enable us to carry out a more precise analysis on the therapeutic efficacy and safety of ethosuximide.
These blood samples will be strictly used in this study, and a specific document informing the patient and obtaining his authorisation will be provided during the first visit (D0-7).
Analgesic and antispasmodic/regulators transit consumption
Drugs consumption for IBS will be evaluated throughout the study during the visits by investigator and throughout the study by the patient on their logbook.
Safety
Any adverse events were collected daily by the patient and during the planned phone calls by investigators (every 7 days). Adverse events are categorised according to their type, intensity and treatment related by investigators. Due to the long half-life of ethosuximide (60 hours), a phone call, within 3 weeks after stopping treatment, will be allowed to recover any adverse effects.
Study discontinuation rate will be evaluated and compared in the two treatment arms.
Data acquisition
The questionnaires will be completed, during each visit, by the patients in the investigating centres. Patients can be assisted in the event of problems understanding the questions. Data collected from the logbook and questionnaires should be verified and indicated on the day of each visit in the electronic case report form (e-CRF). This procedure will allow patients to complete the entire questionnaire and avoid the risk of missing data as much as possible.
Methodology and study design
Study methodology was selected on the recommendation of the European Medicines Agency (ICH guideline Irritable Bowel Syndrome: EMA/CHMP/60337/2013-CPMP/EWP/785/97 Rev. 1; for short treatment on all types of IBS).
Patients will be treated for 12 weeks either by ethosuximide or a placebo, according to a regimen with a specific titration (for details, see figure 1).
Enrolment
Patients followed for the treatment of IBS will be preselected in 10 French centres of academic hospitals. Patients will be contacted in order to briefly present the purpose of the study and make an appointment for the inclusion visit.
Visit 1: inclusion (D0-7) and run-in period (D0-7 to D0)
The objectives of the study, practice organisation and constraints will be explained in details. The investigator will present the information and consent form. The patient must present an IBS diagnostic defined according to the ROME IV criteria. Two blood samples will be taken (blood count, hepatic enzymes, renal function, beta-human chorionic gonadotropin (β-HCG) and genetic analysis).
A daily logbook will be given to the patient with detailed explanations to collect daily, every week before planned visits, their abdominal pain score experienced during the last 24 hours and the Bristol score. Side effects should be collected. The patient will return home with a logbook for 7 days, corresponding to the run-in period. This run-in period, without any studied treatment, was introduced (1) to evaluate the ability of the patients to rate their abdominal pain daily in their logbook and (2) to check the basal average pain intensity over a 1 week period. The patients received the treatment after this run-in period only if the inclusion criteria were met (11-point NRS abdominal pain ≥4 and completed logbook as required by the protocol). The logbook is returned to the patient at each visit. The filled parts will be detached from the logbook and kept on the investigator site in a dedicated binder. The data will then be transcribed as soon as possible on the e-CRF. The data indicated in the e-CRF will not be modifiable and will be confronted with the data indicated in the logbook.
Visit 2: start treatment (D0)
Daily abdominal pain scores have to be filled-in by the patient on the logbook during the last 7 days and the average abdominal pain score should be ≥4 to include the patient in the study. The patients have to fill-in three questionnaires: (1) IBS-SSS, (2) EQ-5D and (3) GIQLI.
If all the inclusion and non-inclusion criteria are conformed, the patient will be enrolled in the study and randomised in one of the two treatment arms (ethosuximide or placebo). The administration dosages, according to a specific titration regimen, will be explained in details to the patient. At the end of the visit, the patient will receive all the therapeutic units for the duration of administration required by the study protocol (4 weeks) and the daily logbook.
Treatment period 1: dose escalation (D0 to D0 +4 weeks)
Ethosuximide or placebo treatment will be administered daily during dinner, according to a regimen of specific titration for 28 days (maximum dose of 10 mL/day). The patient will assess and record the following daily in the logbook:
the evening before bed: abdominal pain felt in the last 24 hours with the 11-point NRS
the evening before bed: the number, shape and consistency of the stool with the Bristol’s scale
side effects or any other particular event should also be carefully noted in the daily logbook
treatment modification.
Every 7 days (at the end of each dose escalation level), patients will be contacted by phone call by the investigator in order to collect information on any side effects and inform the patient of the increased dose in case of inefficiency and good tolerance. Stopping or decrease dosage will be used in cases of intolerance or effectiveness of the treatment.
Visit 3: follow-up visit (D0 +4 weeks)
This first visit will provide information on the therapeutic dose with an optimal benefit–risk ratio for the patient.
During the visit, the patient will complete questionnaires IBS-SSS, EQ-5D, GIQLI questionnaires and the SGA scale. At the end of the visit, the patient will receive all therapeutic units for the next treatment period prescribed by the protocol (4 weeks) and the logbook.
Treatment period 2 and 3 (D0 +4 weeks to D0 +8 weeks and D0 +8 weeks to D0 +12 weeks)
Likewise treatment period 1: to note that the patient will assess and record, only the 7 days before the next visit, the following in the logbook:
the evening before bed: abdominal pain felt in the last 24 hours with the 11-point NRS
the evening before bed: the number, shape and consistency of the stool with the Bristol’s scale.
Visits 4 and 5: follow-up visit (D0 +8 weeks and D0 +12 weeks)
Likewise visit 3: visit 5 closing the treatment period.
To note that at the end of visit 5, will be given to the patient the abdominal pain 11-points NRS to complete daily for 7 days, 3 weeks after the visit 5 (±1 week).
Phone call follow-up to D0 +15 weeks (±1 week)
This phone call aims to recover daily scores of abdominal pain during the last 7 days. This phone call closes the patient’s study participation.
Statistical considerations
Sample size estimation
According to previous work presented in literature,46 we have estimated that a sample size of n=130 patients per randomised group, for a two-sided type I error at 5%, would provide 90% statistical power of detecting an absolute difference of 20% between ethosuximide and placebo in the primary outcome. Based on the meta-analysis of Ford et al, the estimated median responder rate in the placebo group was 35%. A slightly higher rate than the previously mentioned meta-analysis (32% of responders under drug therapies), in order to avoid underestimating the placebo effect. Finally, a total of n=290 patients (145 by group) will be considered to take into account lost to follow-up (10%).
An interim analysis is planned after enrolment of the first 100 patients using the Lan and DeMets rule (East software, Cytel, Cambridge, Massachusetts, USA). The type I error is fixed at 0.03 for this interim analysis. In addition, an early intermediate analysis (third of the inclusions) will be also proposed in order to study safety of the initial maximum dose (10 mL/day) and, if necessary, to allow the addition of a therapeutic dose in case of inefficiency.
Statistical analysis
Statistical analyses will be conducted using Stata software (V.13). A two-sided p value of less than 0.05 will be considered to indicate statistical significance (except interim analysis).
Concerning the primary outcome, the comparison between ethosuximide and placebo will be analysed using χ2 or Fisher’s exact tests. Intention-to-treat analysis will be considered for the primary outcome. Then, the analysis of the primary outcome will be completed by multivariate analysis using a generalised linear mixed model (logistic for dichotomous dependent endpoint) to take into account: (1) fixed effects covariates determined according to univariate results and to clinical relevance (eg, other treatments—analgesic and antispasmodic/regulators transit consumption—gender and IBS subgroup), and (2) centre as random-effects (to measure between and within centre variability). Continuous endpoints (notably abdominal pain intensity) will be compared between ethosuximide and placebo groups using Student’s t-test or Mann-Whitney U test. Normality will be studied by the Shapiro-Wilk test and homoscedasticity using the Fisher-Snedecor test. As suggested by Vickers and Altman,47 the comparison between groups concerning abdominal pain intensity will be completed by linear mixed model considering the 11-point NRS at D0 +12 weeks as a dependent variable and randomisation group and baseline value of pain intensity (D0) as independent parameters (fixed effects). Other categorical parameters (proportion of patients with SGA Scale≥4, IBS severity measured using IBS-SSS questionnaire—mild intensity for a Francis endpoint score <175, moderate for a score from 175 to 300 and severe if the score is >300, analgesic and antispasmodic/regulators transit consumption) and other quantitative outcomes (health related quality of life evaluated using GIQLI and EQ-5D-3L questionnaires) will be analysed as described previously. Longitudinal analyses concerning repeated measures will be studied using random-effect models (linear or generalised linear), to take into account patient as random-effect (slope and intercept), nestled in centre random-effect.
According to clinical relevance and to European Medicines (EMA) and Consolidated Standards of Reporting Trials (CONSORT) recommendations, subgroup analyses depending on IBS subgroup and gender will be proposed after the study of subgroup × randomisation group interaction in regression models (for repeated data or not). Secondarily, a per-protocol analysis will be considered. A descriptive analysis of stopping and discontinuations in view of dose escalation will be considered. A particular focus will be done to safety and lose to follow-up. A study of abandonment considered as censored data will be proposed using the Kaplan-Meier estimation. Finally, a sensitivity analysis will be performed and the nature of missing data will be studied (missing at random or not). According to this, the most appropriate approach to the imputation of missing data will be proposed (maximum bias (eg, last observation carried forward vs baseline observation carried forward) or estimation proposed by Verbeke and Molenberghs for repeated data).
Dissemination
Approval
Any substantial modification of the protocol and of the informed consent form will be presented to the independent medical ethics committee. The latter and the competent French authority will be informed of the end of the study. In accordance with the independent medical ethics committee (CPP Sud Est VI, Clermont-Ferrand, France), no safety and data monitoring committee has been set up in view of the low risk of the ethosuximide treatment. The study is currently registered on the clinical trials website under the following number: NCT 02973542. The protocol has been in V.4 since 28/11/2016.
Patient-informed consent
According to the French law on biomedical research, written informed consent must be obtained from patients prior to participation in the study. Patients will voluntarily confirm their willingness to participate in the study, after having been informed (in writing and verbally) by investigators of all aspects of the study that are relevant to their decision to participate. They will be informed about requirements concerning data protection and have to agree to direct access to their individual data. The patients will be informed that they are free to withdraw from the study at any time at their own discretion without necessarily giving reasons.
Data collection and quality management
A clinical research assistant will be dedicated to data entry, coding, security and storage. Each patient included and the study data will be anonymised. The study data will be collected and managed using an e-CRF (Ennov Clinical, France). A clinical research assistant will be commissioned by the sponsor (University Hospital of Clermont-Ferrand) in order to monitor the progress of the study in accordance with the standard operating procedures implemented in the University Hospital of Clermont-Ferrand, in accordance with good clinical practices and current French laws.
Access to data and dissemination of results
The data set will be the property of the sponsor (University Hospital of Clermont-Ferrand). However, the principal investigator (MD) and the project manager (NK) will have full access to the final data set. The results will be communicated in a peer-reviewed journal, presented at international congresses and completed online on ClinicalTrials.gov.
References
Footnotes
Contributors NK, JS, DA and MD led and contributed to the conceptualisation, design and implementation of this research protocol. BP led the development of the statistical analysis plan. NK participated in the design of the protocol for interventions and assessments. All the authors have read and approved the final manuscript.
Funding This work is supported by funding from the French Ministry for Health (‘Programme Hospitalier de Recherche Clinique Interrégional’, year 2015) and the SFETD (‘Société Française d'Etude et de Traitement de la Douleur’).
Competing interests None declared.
Patient consent Obtained.
Ethics approval The approval of the medical ethics committee (CPP Sud Est VI, Clermont-Ferrand, France) was obtained on 10/10/2016 (ref: AU 1274).
Provenance and peer review Not commissioned; externally peer reviewed.