Article Text

Abstract
Introduction Humeral shaft fractures represent 1%–3% of all fractures and 20% of humeral fractures in adults. The treatment of these fractures is mainly conservative and operative treatment is usually reserved for specific circumstances. To date, no randomised controlled trial (RCT) has compared operative treatment of humeral shaft fractures with conservative treatment.
Methods and analysis We will conduct an RCT to compare the effectiveness and cost-effectiveness of surgical and conservative treatment of humeral shaft fractures. After providing informed consent, 80 patients from 18 years of age with humeral shaft fracture will be randomly assigned to open reduction and internal fixation with locking plate or conservative treatment with functional bracing. We will follow the patients for 10 years and compare the results at different time points. The primary outcome will be Disabilities of Arm, Shoulder and Hand (DASH) at 12 months. The secondary outcomes will include Numerical Rating Scale for pain at rest and in activities, Constant Score and quality of life instrument 15D. Patients not willing to participate in the RCT will be asked to participate in a prospective cohort follow-up study, ‘the declined cohort’. This cohort will be followed up at the same time points as the randomised patients to assess the potential effect of participation bias on RCT results and to enhance the external validity of the RCT. In one of the recruiting centres, all cooperative patients with humeral shaft fractures not eligible for randomisation will be asked to participate in a ‘non-eligible cohort’ study. We will use blinded data interpretation of the randomised cohort to avoid biased interpretation of outcomes. Our null hypothesis is that there is no clinically relevant difference in the primary outcome measure between the two treatment groups. We will consider a difference of a minimum of 10 points in DASH clinically relevant.
Ethics and dissemination The institutional review board of the Helsinki and Uusimaa Hospital District has approved the protocol. We will disseminate the findings of this study through peer-reviewed publications and conference presentations.
Trial registration number NCT01719887; pre-results.
- randomised controlled trial
- humeral fractures
- diaphysis
- shaft
- operative treatment
- functional bracing
- pragmatic cohort
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
- randomised controlled trial
- humeral fractures
- diaphysis
- shaft
- operative treatment
- functional bracing
- pragmatic cohort
Strengths and limitations of this study
Our study is designed to address a common limitation of randomised controlled trials, where only a small subgroup of patients with a specific condition is evaluated and consequently there is a potential for poor external validity.
The study is limited to cooperative patients; thus, certain external validity problems will remain as a significant proportion of these patients are non-cooperative because of multiple trauma, dementia or substance abuse.
Introduction
Background and rationale
Humeral shaft fractures account for 1%–3% of all fractures and 20% of humerus fractures. The overall incidence is around 10–20/100 000 person-years. The incidence increases in elderly patients up to 100/100 000 person-years.1–6 Humeral shaft fractures are mainly caused by simple falls, road traffic accidents and sports injuries.5
The majority of humeral shaft fractures have thus far been treated conservatively using plaster splints, hanging casts or functional bracing.7 8 Operative treatment has generally been reserved to patients with open fracture, multiple trauma, bilateral humeral shaft fracture, concomitant ipsilateral forearm fracture, pathological fracture, or vascular or brachial plexus injury in association with humeral shaft fracture. The most common surgical treatment options are open reduction and internal fixation (ORIF) with plating, minimally invasive plating, and intramedullary nailing or external fixation in severe soft tissue compromise.9–11
Conservative treatment of humeral shaft fracture has been reported to achieve a good outcome.12 13 Sarmiento et al 12 reported a union rate of 97% in their case series of 922 patients, but they were able to follow only 67% of the patients until healing. Zagorski et al 14 reported a 98% union rate and 95% excellent functional results in a case series of 233 patients from the same clinic as Sarmiento et al, but 27% of the patients were lost to follow-up. In case of selectivity in missing data during follow-up, these parameter estimates may be biased and present an overly positive picture of the outcomes of conservative treatment. Recent studies have raised concerns about healing without surgical intervention, especially with certain fracture subtypes. Non-union rates of up to 20%–50% have been reported in proximal and transverse shaft fractures.15–18 Problems with shoulder function after conservative treatment have also been reported.19
Case series of surgical treatment of humeral shaft fractures have reported favourable results with union rates of up to 98%.20–24 Nonetheless, surgical treatment predisposes to certain complications, potentially affecting healing. The reported incidence of non-union ranges from 2% to 10%, infection rates from 2% to 4% and iatrogenic radial nerve palsy from 2% to 7%.22–25 Several trials have compared surgical techniques in treating humeral shaft fractures. According to current literature, ORIF with plating gives comparable or better results in surgical fracture treatment of humeral shaft fractures compared with other surgical treatment options.26–37
The concerns about outcomes from conservative treatment and development in surgical techniques and implants have resulted in a marked increase in the surgical treatment of humeral shaft fractures. There is, however, no high-quality evidence supporting this development.38–40 To date there is no randomised controlled trial (RCT) comparing conservative treatment with operative treatment. This was also noted by a Cochrane database review by Gosler et al.41 We were able to find only one ongoing trial comparing conservative and operative treatment of humeral shaft fractures in a randomised setting.42 We also found one completed and three ongoing prospective observational studies comparing operative and non-operative treatment of humeral shaft fractures.43–46 The use of healthcare services and costs related to different treatment options of the humeral shaft fractures have not been reported in the literature either. We designed the current trial to fill these knowledge gaps.
Objectives and study hypothesis
The objectives of this trial are to compare the effectiveness and cost-effectiveness between surgical treatment with plate osteosynthesis and conservative treatment with functional bracing for humeral shaft fractures.
Our null hypothesis is that there is no clinically relevant difference in the primary outcome measure between the two treatment groups. We will consider a difference of a minimum of 10 points in Disabilities of Arm, Shoulder and Hand (DASH) clinically relevant.47
Trial design
This ongoing trial is a pragmatic, parallel (1:1), multicentre, randomised controlled superiority trial.
Methods and analysis
Study setting
The study is based on a prospective inception cohort design. The randomised trial will be conducted in two university hospitals in Finland (Helsinki University Central Hospital and Tampere University Hospital, with catchment areas of 1.1 million and 0.5 million, respectively) responsible for the treatment of humeral shaft fractures in their respective areas. We will recruit patients for the trial at the trauma centres of the participating university hospitals.
Eligibility criteria
A member of the study group will assess adult patients (18 years or more) with unilateral displaced humeral shaft fractures, referred to the recruiting centre, for eligibility. The diagnosis will be verified using conventional X-ray. If there is a suspicion of extension of the main fracture line either proximally or distally, CT will be used to exclude patients from the trial. Inclusion and exclusion criteria are listed in box 1. All eligible patients will be introduced to the study, given detailed written information about it, asked to participate and to sign the written informed consent form.
Inclusion and exclusion criteria used in the randomised controlled trial
Inclusion criteria
Age: 18 years or older.
Unilateral displaced humeral shaft fracture.
Displacement is at least the amount of the thickness of the cortex, or in transverse fractures diastasis of the half of the thickness of the cortex is required.
The fracture lies in a zone delimited proximally by the superior border of the pectoralis major tendon attachment and distally by the line lying 5 cm from the upper border of the olecranon fossa as evaluated from the X-ray.
The fracture is less than 10 days old.
The operative treatment is performed within 14 days of the trauma.
The patient is willing to accept both treatment options and willing to participate in all follow-up visits.
Patient speaks and reads fluently either Finnish or Swedish (due to language used in data forms).
Exclusion criteria
Bilateral fracture.
Fracture type where pectoralis major and deltoid muscle tendon insertions are in different fracture fragments causing typically significant fracture gap between fragments.
Other concomitant trauma affecting the same upper extremity (fracture, tendon injury, significant soft tissue injury).
Other fracture, thoracic or abdominal injury requiring surgery.
Open fracture.
Pathological fracture.
Polytraumatised patient.
Significant vascular injury.
Plexus injury.
History of trauma of the same upper extremity causing functional deficit.
Trauma or condition that warrants use of walking aid (crutches, wheelchair and so on).
Disease that significantly affects general condition of the patient.
Significantly impaired ability to cooperate for any reason (substance abuse, mental disorder, dementia).
Unwilling to accept both treatment methods.
Interventions
Surgical treatment will be performed either by or under the supervision of an experienced orthopaedic surgeon within 2 weeks after initial trauma by ORIF with 4.5 mm narrow locking compression plate (DePuy Synthes) using either cortical or locking screws and surgical approach (anterolateral, lateral or posterior) preferred by the treating surgeon. The length of the plate will be at least 10 holes to ensure stability of osteosynthesis, and at least three bicortical screws will be used on both sides of the fracture line. Operated patients will be allowed to move their upper extremity without external weight immediately after the operation and gradual weight-bearing will be introduced after 6 weeks.
The control group will be treated with functional bracing (either custom-made brace (figure 1), cork splint, Kir-Fix; or Humerus Comfort brace, NordiCare). The brace will be worn at least 6 weeks or after until objective and subjective union is obtained. Rehabilitation will be started after application of the brace. Active non-weight-bearing exercises of the elbow and hand and pendulum exercises of the shoulder will be allowed immediately. Assisted exercises of the shoulder will be started after 3 weeks and gradual weight-bearing will be added after 6 weeks.

Custom-made functional brace.
All patients will visit a physiotherapist at 3 and 9 weeks for rehabilitation purposes. The rehabilitation protocol is described in table 1.
Rehabilitation protocol
Outcomes
We chose primarily subjective patient-reported outcome measures, since patient experience of the treatment result is paramount in assessing treatment effectiveness. Outcomes will be recorded at 6 weeks, 12 weeks, 6 months and 12 months, 2 years, 5 years and 10 years.
Baseline data
After enrolment, the following baseline data will be gathered: birth date, sex, affected side, dominant hand, fracture type, status of radial nerve, breath alcohol level, injury mechanism, educational background, occupational status and physical workload of current occupation, sporting habits, chronic diseases, medication, previous injuries or diseases of affected upper extremity and treatment received, and smoking habits. Patients will also complete the DASH and the quality of life instrument 15D questionnaires describing the situation prior to the fracture.
Primary outcome
The primary outcome measure of this study is DASH score. The primary time point is at 12 months.
Disabilities of Arm, Shoulder and Hand
DASH is a widely used and validated tool assessing upper-extremity related deficits and symptoms in daily life reported by the patient. It has been shown to be a valid instrument to monitor changes in the symptoms and function over time.47–49 The instrument consists of 30 items and optional items related to work and sports or music (four items each). At least 27 of 30 items must be answered for a score to be calculated. The range of the score is from 0 (no disability) to 100 (extreme disability). DASH is available in several languages, and also in the official languages of Finland (Finnish50 and Swedish51).
Secondary outcomes
Secondary outcomes are both subjective and objective measurements.
A full list of secondary outcomes is shown in box 2. In addition, a questionnaire assessing overall effect of the trauma on daily activities, pain medication usage and patient satisfaction is completed at each follow-up. An X-ray of the affected arm will also be taken at each follow-up.
Outcome measures
Measurements are recorded at 6 weeks, 12 weeks, 6 months, 12 months, 2 years, 5 years and 10 years
Primary outcome measure
DASH at 12 months.
Secondary outcome measures
Pain at rest (0–10 NRS).
Pain at activities (0–10 NRS).
Percentage of patients with acceptable symptom state*.
DASH (other than 12 months).
Constant Score, which includes ROM of shoulder.
ROM of elbow.
Complications.
General satisfaction of the affected shoulder, elbow and the whole upper extremity (0–10 NRS).
General satisfaction of the affected arm and implications in activities of daily living (1–7 Likert scale).
Pain medication usage.
Health service consumption.
Cost-effectiveness.
*See text for definition.
DASH, Disabilities of Arm, Shoulder and Hand; NRS, Numerical Rating Scale; ROM, range of motion.
Numerical Rating Scale for pain
Pain at rest and in activities will be assessed on a 0–10 Numerical Rating Scale (NRS), with 0 (‘no pain’) on the left and 10 (‘worst possible pain’) on the right. NRS is easy to use and is a validated tool for assessing pain.52
Responder analysis
We will carry out a responder analysis, in which the proportions of patients reaching the Patient Acceptable Symptom State (PASS) will be determined. We will analyse the cut-off limit for PASS based on our primary outcome, DASH, using a receiver operating characteristic curve analysis. The patient’s global assessment of satisfaction with the treatment outcome will be used as an anchoring item. Patients who report being very satisfied or satisfied in the patients’ global assessment of satisfaction with treatment outcome will be categorised as ‘responders’.
In addition, we will assess the proportion of patients scoring equal to or less than their preinjury DASH score plus 10 points, which we have chosen as the primary definition of clinical recovery to preinjury level, since the minimal clinically important difference (MCID) of DASH is 10 points.47 However, the DASH score has been shown to have a ceiling effect.53 In a population generally asymptomatic prior to the fracture, the subjects may perceive residual disability with outcomes that fall within the MCID range of the preinjury status of the instrument. Hence, we will also conduct a similar analysis of the patients who reach their reported preinjury DASH score or less, which we consider as a definition of a conservative or a ‘safe’ estimate of recovery to preinjury status.
Constant Score
The Constant Score (CS) is a widely used scale for shoulder function containing both clinician-assessed physical examinations and patient-reported assessments for pain, activities of daily living functions and range of motion (ROM) in the affected and non-affected shoulder.54 We added ROM of elbow to the physical examination, since it has been shown to be affected by humeral shaft fractures.13 ROM is measured with a goniometer and strength with a calibrated spring balance.
15D
The 15D instrument is a widely used generic and standardised self-administered tool used in the assessment of health-related quality of life (HRQoL). The instrument comprises 15 dimensions, and at each follow-up the change compared with the previous follow-up is also recorded. Scoring of the 15D instrument ranges from 1 (no problems in any dimension) to 0 (patient is dead). The results will be used in incremental cost-effectiveness analyses.55
Overall satisfaction
A 7-point Likert scale will be used in the patient questionnaire to describe the overall satisfaction of the situation of the fractured upper extremity and its effect on the patient’s daily living (1=very satisfied, 2=satisfied, 3=somewhat satisfied, 4=not satisfied or dissatisfied, 5=somewhat dissatisfied, 6=dissatisfied, 7=very dissatisfied).
Safety considerations
Potential adverse events will be categorised as serious adverse events (SAEs) and minor adverse events (MAEs). Death, cardiovascular or gastrointestinal events, deep venous thrombosis, pulmonary embolism, secondary permanent radial nerve palsy, deep infection of the fracture site and systemic infections will be categorised as SAEs. MAEs will include, but are not limited to, malunion, non-union, refracture, implant failure, secondary temporary radial nerve palsy and superficial infection of the fracture site. Adverse effects will be registered from the medical records and during follow-up visits. All adverse events will be treated in the study hospitals by or under the supervision of an experienced orthopaedic trauma surgeon.
Anteroposterior and lateral radiographs of the affected arm will be taken at each follow-up visit. The fracture will be deemed non-united if no clinical or radiological consolidation can be seen at 26 weeks after initial trauma. After this time, the patient will be offered an operation to promote healing of the fracture.56
If at any point an imminent problem in healing is observed warranting a change in the treatment regimen, this will be done at the discretion of the treating physician regardless of the initial treatment allocation. This will include, but is not limited to, deep infection, fracture threatening skin integrity, malunion causing subjective problems and a refracture.
Patients having a primary radial nerve palsy will also be included in the study. Current literature suggests that operative exploration is not mandatory in primary radial nerve palsy in association with closed low-energy humeral shaft fracture.57–62 Patients with radial nerve palsy allocated to conservative treatment will be monitored with electromyography at 6 and 12 weeks. If no signs of clinical or electromyographical healing can be seen at 12 weeks, the patient will be offered an elective operation, where affected radial nerve is explored and treated at the discretion of an experienced hand surgeon (neurolysis, nerve repair, nerve grafting, nerve transfer).63
Patients with radial nerve palsy allocated to operative treatment will have the nerve explored in the primary operation and visible lesions will be treated at the discretion of the orthopaedic surgeon. An experienced hand surgeon will be consulted if repair or grafting is necessary. If radial palsy develops after primary operation and there is no mention of the location and status of radial nerve in the operative records, an urgent exploration will be arranged. The function of the radial nerve will be followed with electromyography at follow-up visits. If no healing of repaired, grafted or macroscopically intact radial nerve can be seen 12 months after primary operation, tendon transfer will be offered to regain wrist extension.
Use of healthcare services and cost-effectiveness
In cost-effectiveness analysis, we will consider the consumption of healthcare and social services and related costs including use of pain medication. We will also consider societal costs due to absence from work. Patients will be asked about these subjects in a questionnaire to be completed at every follow-up visit. Consumption of healthcare and social services includes primary healthcare, physiotherapy, occupational healthcare, private and communal outpatient clinics, hospitalisation and alternative medical services (ie, osteopath, chiropractor, naprapath, healer). The costs and benefits will be evaluated against the difference in our main outcomes and in the results of 15D instrument (HRQoL).
Participant timeline
The time schedule of enrolment, interventions, assessments and visits are shown in table 2. A flow chart of the trial is presented in figure 2.
Schedule of enrolment, interventions and assessments

Flow chart of the trial. ORIF, open reduction and internal fixation. IMN, intramedullary nail.
Sample size
The sample size calculation was performed using G*Power 3.164 and was based on DASH as the primary outcome measure in this trial. For the sample size calculation, we used α level of 0.05 and β level of 0.2. We assumed MCID of the DASH to be 10 points, with SD being 14.7.47 65 Using these assumptions, the required sample size is 35 per group with 80% power to show a clinically important difference between the treatment methods with a two-sided type I error rate of 5%. With the assumption of 12.5% lost to follow-up, we decided to include 40 participants per group.
Allocation
Sequence generation and concealment
Sequentially numbered, opaque, identical sealed envelopes were prepared by a statistician with no clinical involvement in the execution of the trial using a computer-generated randomisation schedule. The envelopes were kept in a secure, agreed location at each centre. To minimise the risk of predicting the treatment assignment of the next eligible patient (to ensure concealment), randomisation was performed in unfixed blocks (block size known only to the statistician).
Stratification
Twofold stratification will be used, including status of the radial nerve (intact/paraesthesia/mild motor deficit or subtotal/total motor palsy) and fracture type (AO/OTA type A or type B/C), creating four separate randomisation envelope stacks.
Due to immediate proximity of radial nerve, the overall radial nerve palsy prevalence in patients with humeral shaft fractures was evaluated to be 8%–12%.1 57 Spontaneous recovery of the primary radial nerve palsy has a good prognosis and is not considered an indication for operative treatment.57 60 However, radial nerve palsy may have effect on functional recovery of the upper extremity and was thus taken into account in the stratification.
Fracture type was taken into stratification, since type A fractures are reported to result in non-union more often than type B and C fractures.16
Implementation of randomisation
The allocation sequence was generated by a statistician with no clinical involvement in the execution of the trial. After receiving informed consent, a surgeon member of the study group will open the next sequentially numbered envelope containing the treatment allocation and the surgical treatment or bracing will be arranged accordingly.
Blinding
A physiotherapist unaware of treatment allocation will conduct the objective measurements. The patient will be wearing a long-sleeved shirt and is instructed not to reveal the treatment given when measurements are taken.
Surrounding cohort studies
The generalisability or external validity of RCTs is often discussed and questioned due to the ineligibility of many patients with the condition of interest, or potential participation bias. To enhance the external validity of our trial and to study participation bias, we introduced follow-up cohorts of the patients who decline to participate in the RCT and the patients with a humeral shaft fracture, but are ineligible for the trial.
The declined cohort
Eligible patients unwilling to participate in randomisation will be asked to participate in a follow-up study (referred to as the ‘declined cohort’). Usually, the unwillingness to participate in the RCT is due to a strong preference for one of the treatment modalities and the resulting unwillingness to receive a randomly assigned treatment. The patients will receive usual care with the treatment method (conservative or ORIF) decided by the patient after information on both treatment methods is given. The cohort will follow the same follow-up protocol as the randomised trial (figures 3 and 4). Analysis of the outcome measures will be done separately from the randomised trial, and the results will be compared with the results of the RCT. In addition to controlling for the potential effect of participation bias on external validity, this will provide a possibility to evaluate the potential effects of patient expectations to outcomes, when the patient has been able to participate in the treatment method decision.
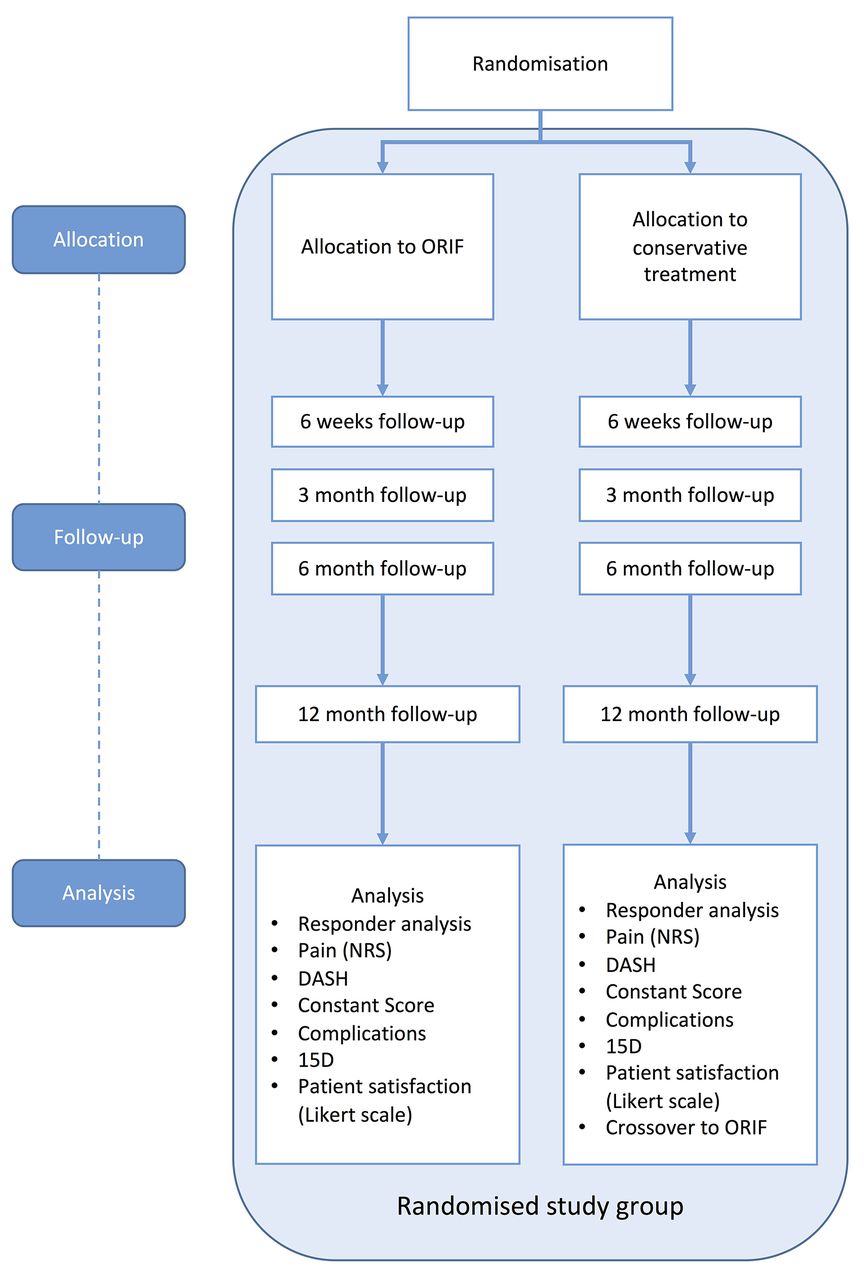
Flow chart of the RCT arm of the trial. DASH, disabilities of arm, shoulder and hand; NRS, numerical rating scale; ORIF, open reduction and internal fixation; RCT, randomised controlled trial.
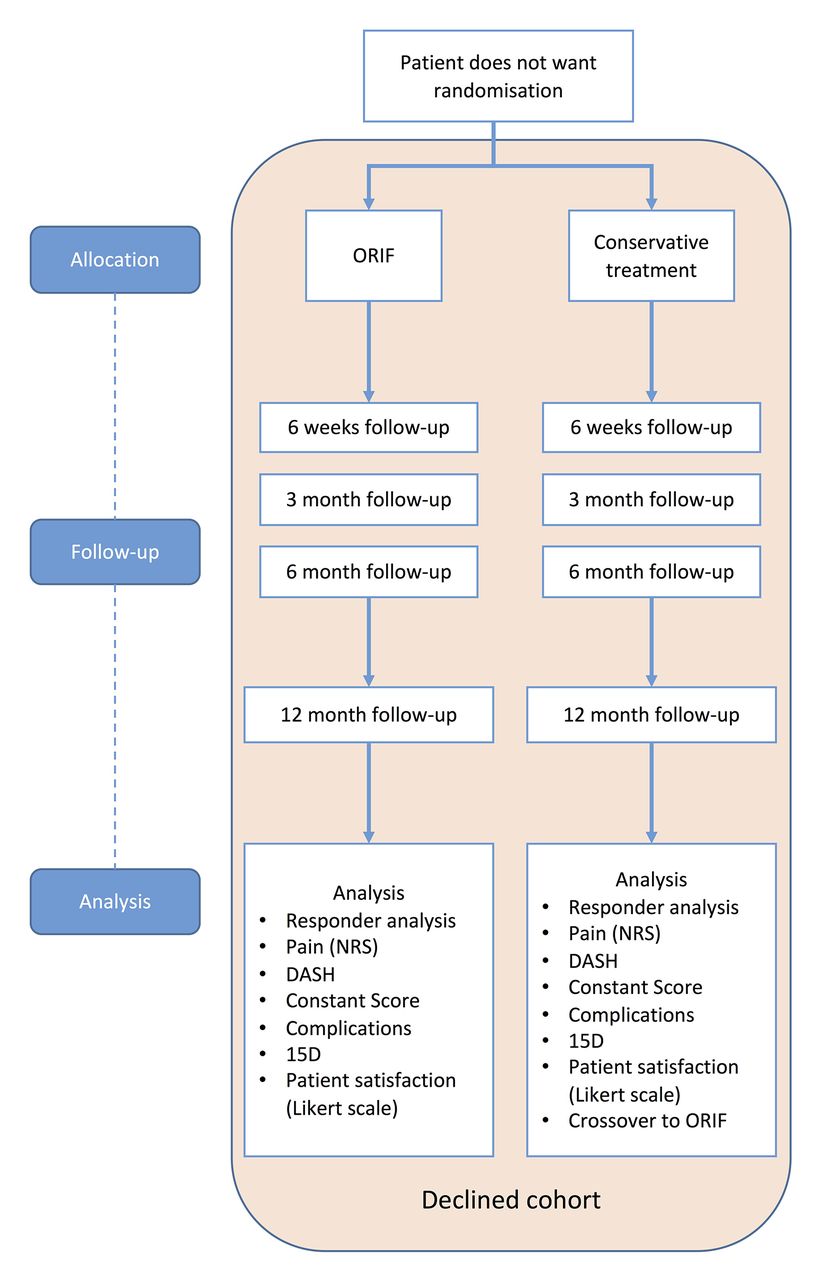
Flow chart of the declined cohort (eligible patients not willing to accept both treatment methods of the trial). DASH, disabilities of arm, shoulder and hand; NRS, numerical rating scale; ORIF, open reduction and internal fixation.
Non-eligible cohort
All compliant patients with fresh humeral shaft fracture, but not eligible for randomisation (the reasons being either fracture extending too proximally or distally, or exclusion criteria are met; see box 1), will be asked to participate in a second prospective cohort study (hereafter referred to as the ‘non-eligible cohort’) in one of the recruiting centres (Helsinki University Hospital). The fracture will be treated at the discretion of treating physician either conservatively or operatively (ORIF or IMN) in the usual fashion. Patients will provide the same baseline data as the randomised patients. The reason(s) for exclusion from randomisation will be recorded and the patient will receive usual care and follow-up visits. The patients will have a study follow-up visit at the outpatient clinic at 12 months, where the same questionnaires and measurements will be performed as in the RCT (figure 5). The results of the non-eligible cohort will give us possibility to enhance the external validity of our trial. Non-compliant patients, fractures older than 2 weeks, end-stage malignancies and patients with such severe trauma that baseline data are not possible to gather (typically patients with multiple trauma with head trauma) will be excluded from the non-eligible cohort.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Flow chart of the non-eligible cohort (cooperative patients not eligible for randomisation). DASH, disabilities of arm, shoulder and hand; IMN, intramedullary nail; NRS, numerical rating scale; ORIF, open reduction and internal fixation.
Data collection and management
Questionnaire forms on paper will be the primary data collection tools for the study. The questionnaires will be completed at the outpatient clinic during the baseline and control visits. On receipt of the questionnaire forms, the researcher will make a visual check of the responses and will query missing data when possible. The paper forms will be securely stored at both study sites.
Double data entry will be used to minimise typing errors. A research nurse and a research assistant will enter the data independently into two separate electronic databases. First, the research nurse enters the data to an electronic database, which is located in a secure network drive and protected with access codes known only by the research nurse. Missing, implausible and inconsistent data in the electronic database will be checked by the research nurse at the coordinating centre. If a missing or implausible item is noticed, the patient will be contacted and asked about the item. The answer will be corrected on the original paper form with a note that the answer was retrieved by a phone call and the corrected data had been entered to the database. Patient records in the participating hospitals will also be used when collecting missing data or interpreting inconsistent or implausible data.
After 12-month follow-up visits are completed and all data stored, a research assistant, not involved in the trial, will enter all the data from the paper forms to a separate database. The two databases will be compared for consistency. Discrepancies will be checked from the original paper forms by a research nurse at the coordinating centre. Final interpretation of the data will be corrected into the master database, which will be the source for the final data analysis.
Participant files will be maintained in storage (both in electronic and paper format) at the coordinating centre for a period of 5 years after completion of the study (10-year follow-up visits).
Missing items
We will use multiple imputation to handle missing data for statistical analyses that cannot handle occasional missing values. All variables that will be included in the final analyses will be included in the chained equations imputation model. The imputation algorithm, fully conditional specification, uses a specific univariate model for each variable and, for each specific imputed data set, iteratively imputes each variable with missing values and uses the imputed values in the imputation of other variables.
Statistical methods
The data will be analysed using IBM SPSS Statistics V.23 or higher. The results will be reported following the Consolidated Standards of Reporting Trials statement.66
The baseline characteristics of the participants will be summarised by group, reported as a mean (SD) or median (first quartile, third quartile) for continuous variables, and count (%) for categorical variables.
Primary statistical analyses will be performed using intention-to-treat basis. For the primary analysis, a mixed-effects model (MM) analysis will be performed using the data set without multiple imputation to compare the mean DASH scores. Treatment group and visits will be included as fixed factors and patient as a random factor. The model will include interactions between treatment and visit. Randomisation stratification factors and baseline value will be included as covariates. The treatment effect will be quantified with an absolute difference between the groups in the DASH score with the associated 95% CI and p value at 12 months postrandomisation.
The MM model will also be used to analyse secondary outcomes where applicable (pain-NRS at rest and during activities, 15D, CS). For categorical response variables, effects will be analysed by logistic regression analysis with treatment as the fixed-factor covariate. These secondary outcomes will only be supportive, explanatory or hypothesis-generating (or both), which is why multiplicity is not considered to be a problem.
The adverse events of the study arms will be reported descriptively. If the number of events is large enough, an analysis between study arms will be performed.
All scale variables will be tested for normality with the Kolmogorov-Smirnov test. Variance of homogeneity will be tested using Levene’s test. We consider a two-sided p value of 0.05 to indicate statistical significance.
We will perform secondary statistical analyses to identify potential effect-modifying and mediating factors. Potential effect-modifying factors to be tested with regression analyses67 are age, gender, body mass index, physical activity, smoking, level of education, fracture of dominant/non-dominant arm and position of the fracture. The absence of adverse effects and treatment attendance as intended will be analysed as a potential effect-mediating factor.
We will also perform an on-treatment analysis if there are patients treated with a non-allocated method because patients declined the allocated treatment after the randomisation, thus causing crossover in study arms. A medical reason to change treatment method, practically from conservative treatment to ORIF because of non-union or fracture threatening skin integrity in the early phase of treatment, will not be considered as a crossover. However, we will analyse such patients in a separate subgroup.
The results of the declined cohort and the non-eligible cohort will also be tested and reported using the statistical methods described above. Differences between the results of different study groups will be tested using the MM model.
Blinded data interpretation
To avoid biased interpretation of the trial data, blinded data interpretation will be used in the reporting of the results of this trial.68 Before accessing the primary outcome data, the Writing Committee will record a ‘Background assumptions’ document, which will contain our definition of MCID of the outcome measures and a brief summary of the key statistical analysis used in the evaluation of the outcome data. The document will be signed by the members of the Writing Committee and published as an appendix to the primary publication. After this, the Writing Committee will write two interpretations of the trial results on the basis of a blinded review of the primary outcome data (treatment A compared with treatment B), with the assumption that A is the ORIF group and another assuming that A is the conservatively treated group. Decisions regarding the key analyses and presentation format for the primary publication before data analysis will also be decided in a meeting of the Writing Committee. The minutes of this meeting will be recorded as a statement of interpretation document, which will be signed by all members of the Writing Committee before the unsealing of the randomisation. This document will be published as an appendix to the primary publication.
Monitoring
Data monitoring
We will conduct the study without a data monitoring committee (DMC). Both treatment methods are widely used in daily practice and have been proven to provide acceptable results. Since there is no DMC, we will not conduct an interim analysis during the trial.
Harms
All the medical records of the participating patients will be carefully assessed, and all harms and complications of the treatment will be reported when reporting the results of this trial. The harms will be categorised to serious and minor adverse events as described in the Safety considerations section.
Auditing
We will not conduct auditing between the participant centres during the trial.
Ethics and dissemination
Research ethics approval
This trial will be conducted according to the Helsinki Declaration. The protocol has been approved by the institutional review board of the Helsinki and Uusimaa Hospital District (approved on 14 May 2012, Dnro 118/13/03/02/2012), and the trial has been duly registered at ClinicalTrials.gov (NCT01719887).
Protocol amendments
All modifications of the study protocol will be communicated by updating the trial registry (ClinicalTrials.gov).
Consent
The informed consent will be obtained by the recruiting doctor at the participating centre. The consent form is written either in Finnish or Swedish and the patient can choose which one to use. The consent forms are supplied as online supplementary files (supplementary materials 1 and 2). Consent will also be obtained from patients belonging to the declined cohort and the non-eligible cohort.
Confidentiality
Both participant data forms and electronic databases will be maintained in secure storage at the coordinating centre for 10 years after completion of the study (after the last patient has reached the 10-year follow-up point).
Access to data
The research nurse in the participant centre is the only person who has access to the electronic trial data during the data collection. After the final data set is formed from the primary data, data set access will be limited to statisticians and the authors of the final publication. The codes of the RCT arms will be known only to the research nurse until the blinded data interpretation has taken place.
Ancillary and post-trial care
Patients will be treated during and after the trial with the best intention. In addition to the planned follow-up visits, patients will receive additional physiotherapy and other interventions at the discretion of the treating doctor. Participating patients will not receive any compensation from the harms of the treatment beyond the compensation from the Finnish Patient Insurance Centre if malpractice has taken place.
Dissemination policy
The findings of this study will be disseminated through peer-reviewed publications and conference presentations. Patients participating in the trial will be sent a letter with information on the results after the primary outcome results are published.
Discussion
In this protocol paper we describe an RCT assessing the effectiveness and cost-effectiveness of surgical treatment of humeral shaft fractures. To date there is no RCT comparing surgical treatment with conservative treatment in humeral shaft fractures. We chose the ORIF with plating as the surgical method in this trial since it is the most widely used surgical treatment method of humeral shaft fractures in Finland. After completion, this trial will provide valuable evidence on the treatment of humeral shaft fractures.
In this trial, we chose primarily patient-reported outcome measures. In older studies of humeral shaft fractures, the outcomes tended to be surgeon-reported, which does not provide insight on how the patient perceives the treatment result. In addition to assessing the percentage of patients with good recovery with a certain treatment, we think it is also important to assess the percentage of poor results achieved with the same treatment, since this permits evaluation of the net benefit of the treatment.69 Recognition of patients with poor results also gives us the possibility to evaluate the risk factors leading to poor results.
Generalisability
RCT is considered the gold standard of scientific evidence for the efficacy of an intervention. When planning an RCT, investigators should consider whether they are performing an efficacy trial to prove that their treatment concept works in ideal circumstances (homogeneous patient material, experienced physicians), or a pragmatic effectiveness trial which tests how the treatment works under usual circumstances (more heterogeneous patient material, variation in the experience of treating physicians). The previous setting usually has a higher internal validity but lower external validity (ie, generalisability), while the latter has better external validity with lower internal validity.70 71
The current trial is designed to be pragmatic effectiveness trial. Nevertheless, a number of patients with humeral shaft fracture will be excluded because of our exclusion criteria and due to the fact that not all patients are willing to accept randomisation. To enhance the generalisability of our trial, we have introduced the declined and non-eligible cohorts as described above. These cohorts will give us the possibility to evaluate the external validity of the randomised group results.
Expectations
We expect operative treatment to result in earlier recovery and more satisfied patients than non-operative treatment at 6 weeks to 6 months, but we expect the differences to be clinically and statistically insignificant at 12 months.
The recruitment began at the end of 2012. At the current recruiting pace, the recruiting will end in 2018.
References
Footnotes
Contributors LR, VL, TL, ST, AM and MP developed the trial, LR being the principal investigator. LR drafted the manuscript and all the members have actively contributed in the further writing of the manuscript. VL is responsible for the trial in Tampere University Hospital. LR, VL, TL and MP are assessing the eligibility and inclusion of the patients to the trial. AM is responsible for the health economic analysis. ST is responsible for the statistical methodology. All authors have read and approved the final manuscript.
Funding This work is supported by The Research Foundation for Orthopaedics and Traumatology in Finland, The Finnish Medical Foundation and University of Helsinki Funds. No direct or indirect funding will be acquired from industry related to this study setting.
Competing interests None declared.
Patient consent No patient pictures or identifiable patient data have been used in this protocol paper. There is one picture of a human being, with custom-made orthosis. This picture is a picture from our clinic and in the picture is one of the authors and a practice model of orthosis was put on him for educational purposes. The consent has been obtained from the person in the picture.
Ethics approval The institutional review board of the Helsinki and Uusimaa Hospital District (Approved on 14 May 2012, Dnro 118/13/03/02/2012).
Provenance and peer review Not commissioned; externally peer reviewed.