Article Text

Abstract
Introduction Within the context of antimicrobial stewardship programmes, de-escalation of antimicrobial therapy is one of the proposed strategies for reducing the unnecessary use of broad-spectrum antibiotics (BSA). The empirical treatment of nosocomial and some healthcare-associated bloodstream infections (BSI) frequently includes a beta-lactam with antipseudomonal activity as monotherapy or in combination with other drugs, so there is a great opportunity to optimise the empirical therapy based on microbiological data. De-escalation is assumed as standard of care for experts in infectious diseases. However, it is less frequent than it would desirable.
Methods and analysis The SIMPLIFY trial is a multicentre, open-label, non-inferiority phase III randomised controlled clinical trial, designed as a pragmatic ‘real-practice’ trial. The aim of this trial is to demonstrate the non-inferiority of de-escalation from an empirical beta-lactam with antipseudomonal activity to a targeted narrow-spectrum antimicrobial in patients with BSI due to Enterobacteriaceae. The primary outcome is clinical cure, which will be assessed at the test of cure visit. It will be conducted at 19 Spanish public and university hospitals.
Ethics and dissemination Each participating centre has obtained the approval of the ethics review committee, the agreement of the directors of the institutions and authorisation from the Spanish Regulatory Agency (Agencia Española del Medicamento y Productos Sanitarios). Data will be presented at international conferences and published in peer-reviewed journals.
Discussion Strategies to reduce the use of BSA should be a priority. Most of the studies that support de-escalation are observational, retrospective and heterogeneous. A recent Cochrane review stated that well-designed clinical trials should be conducted to assess the safety and efficacy of de-escalation.
Trial registration number The European Union Clinical Trials Register: EudraCT number 2015-004219-19. Clinical trials.gov: NCT02795949. Protocol version: V.2.0, dated 16 May 2016. All items from the WHO Trial Registration Data Set are included in the registry.
- De-escalation
- Enterobacteriaceae
- bloodstream infection
- broad-spectrum antibiotics
- antimicrobial stewardship
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
- De-escalation
- Enterobacteriaceae
- bloodstream infection
- broad-spectrum antibiotics
- antimicrobial stewardship
Strengths and limitations of this study
It will be the first trial on de-escalation specifically in patients with bacteraemia due to Enterobacteriaceae.
It will include patients independently of the source of bacteraemia or severity of clinical presentation.
A remote automatic randomisation system and external evaluation by blinded investigators were used to avoid bias.
It has been designed with daily clinical practice in mind.
The open-label design is theoretically more prone to bias.
Switching to oral therapy could potentially reduce the number of days in which patients are assigned to one or other arm.
Background
The worldwide spread of antimicrobial resistance is recognised as a current global public health threat. The implementation of stewardship programmes for optimising antibiotic use has been shown both to improve antibiotic use and also to help combat antimicrobial resistance.1 Streamlining or de-escalation of antimicrobial therapy is a strategy proposed to reduce the unnecessary use of broad-spectrum antimicrobials (BSA).1 2 This can be carried out by changing from combination therapy to monotherapy or by replacing the empirical antibiotic with one with a narrower spectrum of activity, irrespective of the microbiology results.1
Bloodstream infections (BSI) are known to be major causes of morbidity and mortality. They represent suitable organisms for carrying out a de-escalation strategy because they are very frequent, a high proportion of patients are treated with BSA and the susceptibility of the causative organisms is known. The Enterobacteriaceae as a group is the most common cause of community and nosocomial BSI, with a crude associated mortality of around 15%.3 The empirical treatment for nosocomial and some healthcare-associated BSI frequently includes a beta-lactam antibiotic with antipseudomonal activity in monotherapy or in combination. This imposes strong selection pressure, particularly on Pseudomonas aeruginosa isolates, and may be selection of multidrug-resistant Enterobacteriaceae isolates. De-escalation according to microbiological results is assumed as standard of care by most infectologists; however, the reality is that de-escalation is much less frequent than is desirable.4 5 Some of the possible reasons for this phenomenon6–8 include the fact that the safety and efficacy of this treatment strategy are based only on a few observational studies9 10 and expert recommendations.11 12 This was supported by a recent Cochrane review13 conducted among adults with sepsis, severe sepsis or septic shock, whose authors concluded that there is no adequate direct evidence that de-escalation of antimicrobial agents is effective and safe in this scenario. Randomised clinical trials of their safety and efficacy are needed, in order to establish ‘proof of concept’ and help make clinical decisions.
Methods/design
Study hypothesis
The aim of the trial is to demonstrate that de-escalation from empirical therapy with an antipseudomonal beta-lactam to a targeted therapy is as effective and safe in patients with BSI due to Enterobacteriaceae as continuing with the empirical regimen.
Design
The SIMPLIFY trial is a multicentre, open-label, phase III randomised controlled clinical trial, powered to demonstrate the non-inferiority of de-escalation with respect to continuing with the antipseudomonal agent and designed as a real-world pragmatic trial. It was developed in accordance with an extension of the SPIRIT statement for reporting non-inferiority, superiority and equivalence trials.14 15
Participants and settings
The trial will be conducted at 19 public and tertiary Spanish hospitals with the support of the Spanish Network for Research in Infectious Diseases (REIPI) and the Spanish Clinical Research Network (SCReN). Thirteen of them are university hospitals. Patients will be evaluated for eligibility once Enterobacteriaceae is isolated from blood cultures and susceptibility data are available. Detection of eligible patients will be by daily review of blood culture results by infectious disease specialists from the bacteraemia team at each centre. To be enrolled, participants will need to fulfil all inclusion and exclusion criteria (box 1) and give written informed consent (the patient or a legally authorised representative).
Inclusion and exclusion criteria
Inclusion criteria
Written informed consent has been obtained from the patient or the legally authorised representative.
Age ≥18 years, not legally incapacitated.
Hospitalised patients with monomicrobial bacteraemia due to Enterobacteriaceae from any source.
The patient has received active empirical antibiotic therapy with an antipseudomonal beta-lactam (imipenem, meropenem, piperacillin-tazobactam, cefepime, ceftazidime, aztreonam), alone or in combination with another antimicrobial agent, which started in the first 24 hours after the first positive blood culture was taken.
The isolate is susceptible to at least one of the antibiotics included in the experimental arm.
Intravenous antimicrobial treatment is planned for at least 5 days once Enterobacteriaceae is isolated from the blood culture.
Exclusion criteria
Life expectancy <30 days.
Pregnancy or nursing. For included women: failure to use a highly effective contraceptive method.
Isolation of carbapenemase-producing Enterobacteriaceae (because most hospitals do not use monotherapy in these cases).
Inclusion is delayed >48 hours after susceptibility data of the isolate are available.
Severe neutropenia (<500 cells/mm3) on the day of randomisation.
Planned duration of treatment >28 days (eg, osteomyelitis, endocarditis).
Randomisation
Stratified randomisation in a 1:1 ratio will be achieved using a centralised, web-based automated randomisation system integrated with the electronic case report form (eCRF) to manage assignment to the treatment arms. A copy of the randomisation list will be kept in a safe place in case technical problems arise. The only criterion for stratification will be source of BSI (urinary tract vs any other) in order to ensure that the percentage of patients with urinary tract infections is similar in the two groups being compared. To guarantee an appropriate allocation concealment in an open trial, randomisation will not be stratified by site.
Intervention
A decision tree of enrolment to the study is included in figure 1. As stated above, all included patients will already be receiving an antipseudomonal beta-lactam (meropenem, imipenem, piperacillin-tazobactam, cefepime, ceftazidime or aztreonam) before randomisation occurs. Patients will be allocated to one of the following treatment arms:

SIMPLIFY—decision tree of patient enrolment.
Experimental group
The patient will change to an intravenous therapy with an active narrow-spectrum antibiotic according to the susceptibility results (EUCAST or CLSI criteria); the antibiotic will be chosen in the following order (the first active drug will be used): (1) ampicillin, 2 g every 6 hours; (2) trimethoprim/sulfamethoxazole, 160/800 mg every 8–12 hours; (3) cefuroxime, 750–1500 mg every 8 hours; (4) cefotaxime 1–2 g every 8 hours or ceftriaxone, 1 g every 12–24 hours; (5) amoxicillin/clavulanate, 1 g/125 mg every 8 hours; (6) ciprofloxacin, 400 mg every 12 hours; and (7) ertapenem, 1 g every 24 hours. Trimethoprim/sulfamethoxazole will only be used in urinary tract infections in the absence of an undrained renal abscess. Ciprofloxacin is included because, apart from being active against P. aeruginosa, it is not a beta-lactam.
Control group
Continuation of the antipseudomonal beta-lactam was being administered on an empirical basis: meropenem, 1–2 g every 8 hours; imipenem, 0.5–1 g every 6 hours; piperacillin-tazobactam, 4/0.5 g every 6–8 hours; cefepime, 2 g every 8–12 hours; ceftazidime, 1–2 g every 8 hours; and aztreonam, 1–2 g every 8 hours.
Exceptions to the above rule
Third-generation cephalosporins should be avoided where there are inducible AmpC beta-lactamase-producing Enterobacteriaceae (Enterobacter spp, Providencia spp, Morganella morganii, Serratia marcescens and Citrobacter freundii); hence, even if the isolates are strictly susceptible, for patients in the control group, ceftazidime may be changed to any other antipseudomonal beta-lactam on the day of randomization. For patients allocated to the experimental arm, the options will be limited to trimethoprim/sulfamethoxazole, ciprofloxacin or ertapenem.
Extended-spectrum beta-lactamase (ESBL) producers could be included in the study based on attending physician’s criteria; in these cases, maximum doses of susceptible antibiotics are recommended.
Dose adjustment
Due to the nature of the study design as a real-world clinical practice trial, antimicrobial dosage will be as deemed by the treating clinician, dependent on pharmacokinetic and pharmacodynamic (PK/PD) characteristics (such as higher doses for septic shock or high body mass). Dose adjustment will be made for all drugs as necessary in the case of renal or hepatic dysfunction, following summary of product characteristics recommendations.
Concomitant therapy
Even if the BSI is monomicrobial, the attending physician may consider the infection to be polymicrobial at source. If additional anaerobic or Gram-positive coverage is needed, concomitant use of oral metronidazole, clindamycin, vancomycin, teicoplanin, daptomycin or linezolid is allowed in both arms. Concomitant treatment with any other systemic antibiotic with intrinsic activity against Gram-negative bacilli is not allowed. The administration of any of these drugs while the patient is receiving the study drug will be deemed a criterion for withdrawal. There are no absolute contraindications for the use of any other drug during the study. However, contraindications, warnings and precautions for use and possible interactions with study drugs are to be taken into account.
Duration of therapy
The appropriate duration of therapy is considered to be between 7 and 14 days, according to the attending physician’s criteria. Treatments lasting longer than 14 days will be allowed only when there is an undrained abscess present, in which case, a 4-week treatment is permitted.
Route of administration
Switching to oral therapy is allowed after the third day of therapy after randomisation if all the following conditions are fulfilled: clinical improvement has been achieved, absence of fever (>38°C), haemodynamic stability, adequate control of the source of BSI and absence of secondary foci, adequate oral intake, and no gastrointestinal conditions that might compromise drug absorption.
For patients in the experimental group, switching to oral therapy is allowed with the same intravenous drugs as follows: trimethoprim/sulfamethoxazole 160/800 mg every 8–12 hours, cefuroxime axetil 500 mg every 8–12 hours, amoxicillin/clavulanate 875/125 mg every 8 hours or ciprofloxacin 500 mg every 12 hours. If the intravenous drug is ampicillin, amoxicillin 1 g every 8 hours will be used; if cefotaxime or ceftriaxone, then ceftibuten 400 mg every 12–24 hours or cefixime 400 mg every 12–24 hours will be used; if ertapenem, this drug may be switched to the intramuscular route.
For patients in the control group, the preferred oral option is ciprofloxacin 500 mg every 12 hours for all patients. The protocol allows treatment with cefuroxime-axetil 500 mg every 8–12 hours or cefixime 400 mg every 12–24 hours only in cases of resistance to ciprofloxacin; finally, parenteral ertapenem 1 g every 24 hours may be used for convenience if the isolate is resistant to all other oral options.
Rescue medication
No rescue medication is planned on behalf of the study if a patient has to withdraw from the trial for any reason; the attending physician will follow clinical guidelines for routine clinical practice and Good Clinical Practice rules.
Medication
As all the study drugs are recommended for BSI caused by Enterobacteriaceae, the sponsor will not provide the study drugs.16 Every site participating in the study is authorised to use the drugs through the normal provision of its hospital pharmacy.
Schedule of visits
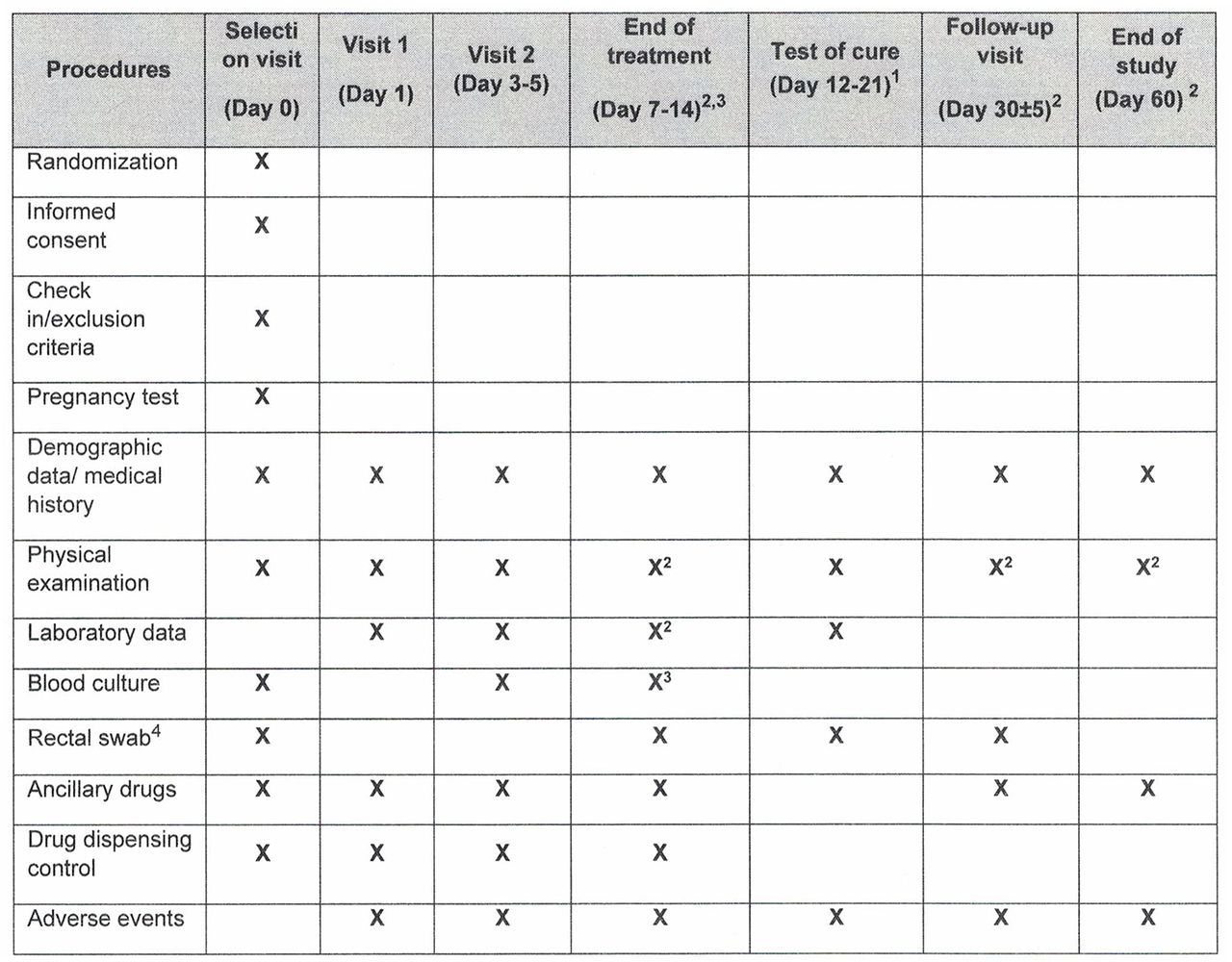
Patients included in the study will be followed for 60 days (±5 days) after diagnosis of the BSI (figure 2). Follow-up will be organised in seven planned visits at day 0 (baseline), day 1; days 3–5; days 7–14 (end of treatment); days 3–5 from end of treatment (test of cure, TOC); day 30±5; and day 60±5. Visits at days 30 and 60 may be made by telephone.
{kind=link}
{kind=link}
Schedule of visits and assessments. Except where otherwise specified, these refer to days from randomisation.
The visit schedule is planned so as to obtain data on clinical status, sample collection, efficacy and safety variables, and adverse events. At the final evaluation at 60 days, data on all outcome variables will be gathered. Additionally, data will be collected at unplanned visits, with special consideration given to the occurrence of any adverse event or recurrence.
Outcomes
The primary outcome is clinical cure, which will be assessed at the TOC visit (3–5 days after the end of antibiotic treatment). Death during treatment, change of antibiotic therapy due to clinical failure or need to prolong the treatment will be considered as failures (table 1). Secondary outcomes include early (5 days after end of treatment) and late (60 days) clinical and microbiological response, all-cause mortality (days 7, 14 and 30), length of hospital stay, recurrence rates (relapse or reinfection) (day 60), safety of antibiotic treatment, including Clostridium difficile infections and number of antibiotic treatments with an antipseudomonal beta-lactam; in a subgroup of patients, the rate of intestinal colonisation by P. aeruginosa resistant to carbapenemase or piperacillin/tazobactam, Stenotrophomonas spp, multiresistant A. baumannii and enterobacteria producing ESBL, carbapenemase and chromosomal AmpC (hyperproduction) or plasmid will be sought. Some of these secondary outcomes will be analysed as composite variables, following the Desirability of Outcome Ranking (DOOR)/Response Adjusted for Duration of Antibiotic Risk (RADAR) methodology. Outcome definitions, assessment and time frames for measurement are described in table 1.
Outcome definitions and time frames
Data collection, management and monitoring
The coordinating centre for this study is the Hospital Universitario Virgen Macarena, Seville, Spain, and the Clinical Trials Unit (CTU-Hospital Universitario Virgen del Rocío) has delegated sponsor functions on behalf of the Fundación Pública Andaluza para la Gestión de la Investigación en Salud de Sevilla (http://www.fisevi.com). Clinical research associates connected to the SCReN in public hospitals will carry out monitoring activities. Data collection will be conducted by trained staff at each participating centre and entered into a restricted access eCRF. These forms will be available at the eCRF web platform. Outstanding queries regarding the completion of the CRF will be sent to all participating centres as necessary to ensure accuracy of data.
In order to avoid any association with personal data, all study samples will be anonymous and identifiable only by the patient’s alphanumeric study code. The objective and management of these samples are included in the patient’s information sheet and informed consent form.
The quality of all data collected will be carefully supervised by the CTU, and specific visits for source data verification are organised according to the monitoring plan. Furthermore, in order to minimise bias, at the interim analysis (when 50% of the sample has been included), an independent committee (three independent investigators from the REIPI) blinded to treatment assignment will review all accumulated data. This committee will advise the sponsor on the appropriateness of continuing the clinical trial as designed.
Isolates
All isolates will be sent to the central laboratory in the Hospital Universitario Virgen Macarena in Seville for susceptibility testing using reference methods and PCR characterisation and sequencing if necessary.
Eight selected hospitals will participate in the study of rectal carriage of ESBL-AmpC- and carbapenemase-producing Enterobacteriaceae, by taking rectal swabs from participants at different times (as set out in the schedule of visits). To do this, samples will be taken by rectal swab from the patients of both treatment arms on the day of randomization, the day when treatment finish, the day of test of cure and visit of day 30. The presence of P. aeruginosa resistant to carbapenemase or piperacillin/tazobactam, Stenotrophomonas spp, multiresistant A. baumannii and enterobacteria producing ESBL, carbapenemase and chromosomal AmpC (hyperproduction) or plasmid will be sought. A written consent form for samples, approved by the ethics committees, is also provided for the study.
Definition of analysis population and outcome measures
The following populations will be considered: the intention-to-treat population (ITTP) includes all randomised patients; the modified ITTP (mITTP) includes randomised patients who have received at least one dose of intravenous antibiotics; the clinically evaluable population (CEP) includes patients who have completed 5 days of the intravenous study drug or who die but have received at least one dose of intravenous antibiotics. The clinically and microbiologically evaluable population (CMEP) includes those in the CEP who have had microbiological tests (at least one blood culture 48 hours after randomisation).
The local principal investigator in the centre where the patient was included will assess the primary outcome (clinical cure) in the CEP at TOC. Due to the intrinsic characteristics of the primary outcome (soft outcome) and the study methodology (non-blinded), this evaluation done will be reviewed later on the basis of clinical data recovered on two occasions by an external blinded investigator: first, during the interim analysis to monitor safety; second, before the complete cleaning and closure of the eCRF. For secondary endpoints, the CMEP will be eligible for early (day 5) and late (day 60) microbiological responses, the m-ITTP for all-cause mortality and length of hospital stay, and the CEP for the evaluation of recurrence rates and drug safety.
Sample size
The sample size was calculated using Epidat V.4.0. Some of the data used to calculate it was derived from the study published by Retamar et al.17 Assuming estimated clinical cure rates of 85% in both groups, a non-inferiority margin of 10% difference between the two groups and treatment assignment in a 1:1 ratio, 344 patients in total (172 per study arm) are needed to achieve 80% power with a significance level of 5%. This allows for a 5% drop-out rate. The 10% non-inferiority margin was chosen as in recent trials of complicated urinary tract and intra-abdominal infections.18 19
Statistical analysis
Absolute differences will be calculated with 95% CIs for the clinical cure rate between the two arms of the study at TOC. Multivariate analysis using logistic regression for the main outcome will be performed in order to ensure the independence of the treatment effect. Special consideration will be given in the multivariate analysis to the centre of origin of the study sample. A Cox regression analysis of mortality until 60 days will be performed on the mITTP. For the superiority analysis, logistic regression will be used sequentially, using the methodology recently published by Evans et al 20 for the composite variable (DOOR and RADAR analysis using survival at day 14, number of days of antipseudomonal beta-lactam treatment avoided, presence or absence of side effects, including C. difficile infections, secondary MDRO infections, and all drug-related adverse events). Antimicrobial doses are not fixed, and sensitivity analyses will therefore be applied to control potential bias.
Protocol violations
All protocol violations occurring after randomisation will be listed in the clinical study report, tabulated by subject and by recruitment centre.
Ethics and dissemination
Each of the participating centres has obtained the approval of an ethics review committee, the agreement of the directors of the institutions (who signed the contract of agreement with the sponsor of the study) and authorisation from the Spanish Regulatory Agency (Agencia Española del Medicamento y Productos Sanitarios). All the patients have to sign the informed consent previous to the randomisation (see online Supplementary data). Patients may withdraw from the study at any time without prejudice, as is documented and explained at the time of providing consent. The study will be carried out according to the principles of the Declaration of Helsinki and Directive 2001/20/EC of the European Parliament and of the Council of 4 April 2001 on the harmonisation of the laws, regulations and administrative provisions of the member states relating to the implementation of Good Clinical Practice in the conduct of clinical trials on medicinal products for human use until the new Clinical Trials Regulation EU No 536/2014 becomes applicable, which will be no earlier than 28 May 2016. The confidentiality of records that might identify subjects in this study will be protected in accordance with EU Directive 2001/20/EC. All laws for the control and protection of personal information will be carefully followed. The identities of patients will not be disclosed in the eCRF; names will be replaced by an alphanumeric code, and any material related to the trial, such as samples, will be identified in the same way, so that no personal information will be revealed.
Regarding to the dissemination plan, three communications with preliminary clinical data to national and international conferences (American Society for Microbiology/Infectious Diseases Society of America or European Congress of Clinical Microbiology and Infectious Diseases) are proposed during the second year of the study. For the third year, a further presentation will be given at a national conference and two other presentations at international conferences with final or advanced data. Once we obtain the final results of the study, at least three publications are expected: one in a D1 journal and two in Q1 journals.
Discussion
The extensive use of BSA and the dramatic increase in infections due to multidrug-resistant organisms are forcing the scientific community to look for strategies to combat this situation. In the real world, the application of de-escalation to serious infections is less frequent than is desirable. The arguments against de-escalation include (1) the minimum inhibitory concentration of some narrow-spectrum drugs are closer to susceptibility breakpoints than carbapenems, for example, and some physicians may therefore feel safer using the latter; (2) subpopulations resistant to narrow-spectrum drugs may be selected and appear after some days of empirical treatment, leading to treatment failure in case of de-escalation; (3) in the case of polymicrobial infections, it is not uncommon for only one of the pathogens to be isolated in blood cultures, so that simplification of treatment may be less safe and effective than a broad-spectrum treatment; (4) there is some doubt about the real effectiveness of certain drugs against isolates producing specific mechanisms of resistance. Furthermore, although it is assumed that BSA has a greater impact on the selection of multidrug-resistant strains, some studies suggest that it may depend more on the duration of the treatment than the spectrum.16 While none of these arguments have been proven, it is also true that there is no strong evidence for the safety of de-escalation strategies in these scenarios.
To the best of our knowledge, three randomised trials on de-escalation strategies, none of them specifically focused on bacteraemia, have been published, which show significant differences from this study.21–23 The one published by Falguera et al 22 compared the efficacy of empirical versus targeted treatment on the basis of urine antigen results in hospitalised patients with community-acquired pneumonia. The article published by Kim et al 23 evaluated the efficacy of early use of imipenem/cilastatin and vancomycin followed by de-escalation versus conventional antimicrobials without de-escalation for patients with hospital-acquired pneumonia in intensive care units (ICUs). The last one, published recently by Leone et al,24 included a limited number (n=116) of ICU-admitted patients with severe sepsis; its primary outcome was duration of ICU stay and not effectiveness of both treatment strategies. In that study, de-escalation followed the recommendations of guidelines, not a prespecified protocol based on the clinical impact of the antibiotics. There was no significant difference in mortality, although unexpectedly, patients in the experimental arm had a higher rate of superinfections (27% vs 11%; p=0.03). These results contrast with a recent systematic review and meta-analysis that included 25 studies with data on de-escalation based on culture results, which showed a significant reduction in the relative risk (RR) of death (RR 0.44, 95% CI 0.30 to 0.66; p<0.0001). It is important to note that many of the included studies in the meta-analysis were observational, were retrospective and had a high degree of heterogeneity.25
Several authors have warned about the considerable inconsistencies in definitions of de-escalation. In 2015, Weiss et al 26 elaborated a consensual definition of de-escalation that allowed beta-lactams to be ranked according to both their spectra and their ecological impact. The authors underlined the difficulty of reaching consensus on the relative ecological impact of each individual drug. In 2014, Madaras-Kelly et al 27 used the Delphi approach to develop an antibiotic spectrum score to measure de-escalation. We shall therefore include both concepts in our analysis, using Outcome Ranking (DOOR) and Response Adjusted for Duration of Antibiotic Risk (RADAR) analyses.20
Switching from intravenous to oral therapy as soon as the patient is clinically stable can reduce the risk of adverse events related to intravenous therapy, length of hospitalisation and cost. It can be applied regardless of the source of infection and underlying conditions whenever a good option that achieves the PK/PD targets is available.28 In our study, switching to oral therapy is allowed in both arms to avoid exposing patients in the control arm to unnecessary risks.
The SIMPLIFY trial has several strengths. In the first place, it will be the first trial on de-escalation specifically in patients with bacteraemia due to Enterobacteriaceae. Second, it will include patients independently of the source of bacteraemia or severity of clinical presentation. Third, it was designed with daily clinical practice in mind. We hope that, if there is reasonable evidence to reject the null hypothesis, it will encourage implementation of this type of strategy in daily practice.
Trial status
Funding for the study was approved on 15 August 2015 and available for study expenses in 1 January 2016.
Ethics committee approval for the 19 sites included was obtained on 15 March 2016.
Authorisation from the Spanish Regulatory Authority was obtained on 18 March 2016.
The study has been approved for a recruitment period of 2 years.
Dissemination of results directed to patients will be channelled through the Spanish Clinical Studies Registry (Agencia Española del Medicamento y Productos Sanitarios), whose content is adapted to patients.
References
Footnotes
Contributors JR-B and LEL-C were responsible for formulating the overall research questions and for the methodological design of the study. CR-F, BA and LL-A collaborated in the submission of the project for the Spanish funding and collaborated in the methodological aspects of the study. JR-B is the coordinating investigator and leader of the coordination team. CR-F is responsible for the CTU. MN-N collaborated with writing of the manuscript and with the pharmacovigilance design, and JB-F, PR-G and CL collaborated in the organisation of the study. MD contributed in all the microbiological details of the study. JR-B and LEL-C participated in its design and supervised the project. All authors read and approved the final manuscript.
Funding Fundación Pública Andaluza para la Gestión de la Investigación enSalud de Sevilla (FISEVI), Contact: claram.rosso.sspa@juntadeandalucia.es. This project is a non-commercial, investigator-driven clinical trial, funded through public competitive call by the Instituto de Salud Carlos III (ISCIII), document number: PI15/00439, funded by Instituto de Salud Carlos III, integrated in the national I+D+i 2013-2016 and co-funded by European Union (ERDF/ESF, “Investing in your future”). This study is supported by the Spanish Clinical Research Network and funded by ISCII: study number 16.001.
Disclaimer The sponsor and funders of the study had no role in the study design or in manuscript development.
Competing interests None declared.
Patient consent Detail has been removed from this case description/these case descriptions to ensure anonymity. The editors and reviewers have seen the detailed information available and are satisfied that the information backs up the case the authors are making.
Ethics approval Spanish Regulatory Agency.
Provenance and peer review Not commissioned; externally peer reviewed.
Collaborators P Aguilar, IJ de la Calle and A Romero (Hospital Universitario Puerto Real, Cádiz); E Merino, V Boix, L Giner, JC Rodríguez and A Gimeno (Hospital Universitario de Alicante); F Guerrero-Sánchez, A Martín-Aspas and F Galán-Sánchez (Hospital Universitario Puerta del Mar, Cádiz); D Diez, V Pérez-Carral and MI Paz (Complexo Hospitalario Universitario de Ourense); C Fariñas, C Armiñanzas, C González and C Ruiz de Alegría-Puig (Hospital Universitario Marqués de Valdecilla, Santander); L Gómez, E Calbo and M Xercavins-Valls (Hospital Universitario Mutua Terrassa, Terrassa); F García-Colchero and M Chávez (Hospital San Juan de Dios, Sevilla); J Goikoetxea-Agirre, M Montejo and L López (Hospital Universitario de Cruces, Barakaldo); G Bou and I Torres (Hospital Universitario A Coruña); S Pérez-Cortés and MD López-Prieto (Hospital Universitario de Jerez); B Loeches and M Romero (Hospital Universitario La Paz, Madrid); M Ibarguren, MA Goenaga-Sánchez and JM García-Arenzana (Hospital Universitario Donostia); JR Yuste and J Leiva-León (Clínica Universitaria de Navarra); A Salas-Aparicio and C de las Cuevas (Hospital Universitario La Princesa, Madrid); JM Guerra-Laso and I Fernández-Natal (Complejo Asistencial Universitario de León); MT Pérez and F Vasallo (Xerencia de Xestión Integrada de Vigo); G Cuervo and C Ardanuy (Hospital Universitario de Bellvitge); JR Paño and S Salvo (Hospital Clínico Universitario Lozano Blesa, Zaragoza); ML Martín-Pena and E Ruiz de Gospegui (Hospital Universitario Son Espases); M del Barrio, S Sadyrbaeva, M Coronel-Janeiro, MP Alarcón-González and A González-Herrero (Hospital Universitario Virgen Macarena).