Article Text

Abstract
Introduction In spite of advances in understanding and technology, postoperative pain remains poorly treated for a significant number of patients. In colorectal surgery, the need for developing novel analgesics is especially important. Patients after bowel surgery are assessed for rapid return of bowel function and opioids worsen ileus, nausea and constipation. We describe a prospective, double-blind, parallel group, placebo-controlled randomised controlled trial testing the hypothesis that a novel analgesic drug, VVZ -149, is safe and effective in improving pain compared with providing opioid analgesia alone among adults undergoing laparoscopic colorectal surgery.
Methods and analysis Based on sample size calculations for primary outcome, we plan to enrol 120 participants. Adult patients without significant medical comorbidities or ongoing opioid use and who are undergoing laparoscopic colorectal surgery will be enrolled. Participants are randomly assigned to receive either VVZ-149 with intravenous (IV) hydromorphone patient-controlled analgesia (PCA) or the control intervention (IV PCA alone) in the postoperative period. The primary outcome is the Sum of Pain Intensity Difference over 8 hours (SPID-8 postdose). Participants receive VVZ-149 for 8 hours postoperatively to the primary study end point, after which they continue to be assessed for up to 24 hours. We measure opioid consumption, record pain intensity and pain relief, and evaluate the number of rescue doses and requests for opioid. To assess safety, we record sedation, nausea and vomiting, respiratory depression, laboratory tests and ECG readings after study drug administration. We evaluate for possible confounders of analgesic response, such as anxiety, depression and catastrophising behaviours. The study will also collect blood sample data and evaluate for pharmacokinetic and pharmacodynamic relationships.
Ethics and dissemination Ethical approval of the study protocol has been obtained from Institutional Review Boards at the participating institutions. Trial results will be disseminated through scientific conference presentations and by publication in scientific journals.
Trial registration number NCT02489526; pre-results.
- PAIN MANAGEMENT
- CLINICAL PHARMACOLOGY
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Strengths and limitations of this study
This is a prospective, randomised, double-blind design that will evaluate for a clinically meaningful response to a novel analgesic agent. The new drug could broadly expand treatment options for postoperative pain for patients.
The trial assesses for many of the common potential confounders of analgesic response, including perioperative anxiety, depression and catastrophising behaviours that may influence reporting of pain levels and affect use of opioids in the postoperative period.
The protocol includes an assessment of perception of treatment both on the part of participants and investigators, which will help evaluate the effectiveness of blinding to treatment allocation.
A potential limitation is that a diverse and heterogeneous group of patients may be enrolled, potentially confounding the results.
Introduction
Although there have been numerous studies published regarding the use of existing drug classes for analgesia, manuscripts related to opioid-type drugs dominate.1 In spite of significant research efforts, novel non-opioids or non-non-steroidal anti-inflammatory drugs (NSAIDs) have been found to have questionable efficacy in the treatment of postoperative pain, and are not in routine clinical use. As noted by Kissin2 in 2010, there has been no new analgesic drug based on a novel pain mechanism introduced to the market in more than a generation. Therefore, there is an unmet need for such research to come to fruition, especially since postoperative pain continues to be inadequately managed in some patients even with full availability of existing therapies.
The currently available analgesic therapies for acute postoperative pain based on providing opioid analgesics often lead to unsatisfactory pain relief or excessive side effects.3 Providing additional opioids can result in intolerable side effects such as nausea, pruritus, constipation, respiratory depression, and may induce tolerance, rendering the analgesic treatment less effective. In addition, opioid management can lead to the development of hyperalgesia (increased pain sensitivity) in some patients in the postoperative period.4 Furthermore, the use of NSAIDs can be linked to the development of gastrointestinal complications and renal injury, especially in patients who have altered fluid shifts in the perioperative period.5 ,6 Although patients may benefit from perioperative use of drugs like gabapentin, non-opioid, non-NSAIDs are poorly effective as stand-alone agents for acute postoperative pain management.7 Finally, regional anaesthetic techniques, although useful for many patients, also have limitations, side effects, and can still be poorly effective.8
In colorectal surgery, the need for novel analgesics is especially important. Patients after bowel surgery are assessed for rapid return of bowel function and opioids can worsen or prolong ileus, nausea and constipation—which are highly undesirable outcomes in this patient population. Excessive pain and use of opioids not only delays return of normal gastrointestinal function, but patients may require prolonged stays in the hospital—increasing the risk of iatrogenic infections. Prolonged immobility due to pain can increase the risk of thromboembolic events and result in muscle wasting and debilitation, which may have an overall adverse effect on patients' outcomes, satisfaction and quality of life.
Therefore, the need to develop alternate and novel analgesics is desirable for improving management of postoperative pain, being specifically desirable and needed in managing patients postoperatively who have undergone bowel surgery.
VVZ-149 is a small molecular compound in the benzamide family that has been developed as an injectable product to improve postoperative pain control. A novel analgesic drug VVZ-149 is a dual antagonist of glycine transporter type 2 (GlyT2) and serotonin receptor 2A (5HT2A). GlyT2 blockage increases inhibitory synaptic transmission by glycine in the spinal cord, resulting in a reduction of pain transmissions to the brain.9–14 5HT2A blockage decreases descending serotonergic facilitatory modulation on pain transmission by the brain and reduces nociceptor activation in peripheral nerves, both of which are primary sources of postsurgical pain.15–19 There is good rationale for developing drugs that act on multiple targets, as this may improve therapeutic benefit while reducing side effects of therapy.20 VVZ-149 has comparable efficacy to morphine in well-controlled (blind, complete randomisation with a positive control) animal studies using rat models of postoperative pain and formalin-induced pain. A clinical phase 1 study performed in healthy participants has shown no clinically significant adverse events.21
Laparoscopic colorectal surgery is typically associated with a burst of moderate-to-severe pain (Numerical Pain Rating Scale (NRS) ≥4, scale 0–10) in the immediate postoperative period.22 Because of the significant analgesic requirements for patients who undergo this type of surgery, using this postoperative pain model is an appropriate condition under which to study VVZ-149. In addition, due to potentially undesirable effects that opioids often have on bowel function and recovery, studying the efficacy and side effect profile of VVZ-149 is ideally suited to the colorectal surgery patient population.
In summary, VVZ-149 is potentially a favourable non-opioid and non-NSAID analgesic candidate with comparable efficacy to morphine that may be able to provide superior analgesia and reduce side effects for patients who are recovering from laparoscopic colorectal surgery.
Objectives
The overall objective of this multicentre, double-blind RCT is to evaluate the safety and efficacy of VVZ-149 for treating postoperative pain in patients who undergo laparoscopic colorectal surgery. When compared with providing patients with intravenous (IV) opioids alone (hydromorphone patient-controlled analgesia (PCA)), we hypothesise that patients randomised to receive VVZ-149 with hydromorphone PCA will show significantly greater improvement in pain with an acceptable side effect profile compared with patients randomised to receive IV hydromorphone PCA only. We are also examining whether VVZ-149 is effective in reducing opioid consumption, as well as its side effect and safety profile (eg, nausea and vomiting, sedation, ECG changes, other laboratory parameters, and respiratory depression). We are examining potential confounders, such as pre-existing anxiety, depression, and catastrophising behaviours, and reporting on overall patient satisfaction with this novel therapy. Finally, we are also evaluating the robustness of our blinding by assessing expectation of treatment in study participants and perception of treatment in study personnel and patients.
Methods and study design
The VVZ-149 study is a randomised, double-blind, parallel group, placebo-controlled study to evaluate the efficacy and safety of the analgesic drug candidate VVZ-149 for participants with pain following laparoscopic colorectal surgery. Patients, study investigators and the research team collecting data are blind to patient treatment allocation.
Participants of age 18–70 undergoing laparoscopic colorectal surgery are screened within 30 days prior to the day of the surgery. After completion of the surgery, participants are transferred to the postoperative anaesthesia care unit (PACU), where the study drug is initiated as soon as possible once the participant is admitted. During surgery, eligible participants are randomised in a 2:1 ratio to one of two study groups, VVZ-149 injections or placebo. Then, participants who have a pain score of at least 4 on the 11-point NRS are administered the study drug. Participants who are deemed ineligible have no further data collected and receive routine postoperative care.
A randomised participant receives a dosing regimen of VVZ-149 injections or placebo via IV infusion for 8 hours. In addition to receiving the study drug or placebo, all participants receive IV PCA with hydromorphone on demand to facilitate achieving adequate pain relief. Participants are evaluated per the study protocol through 24 hours after dosing. A follow-up assessment is performed 14–30 days following the treatment (figure 1).
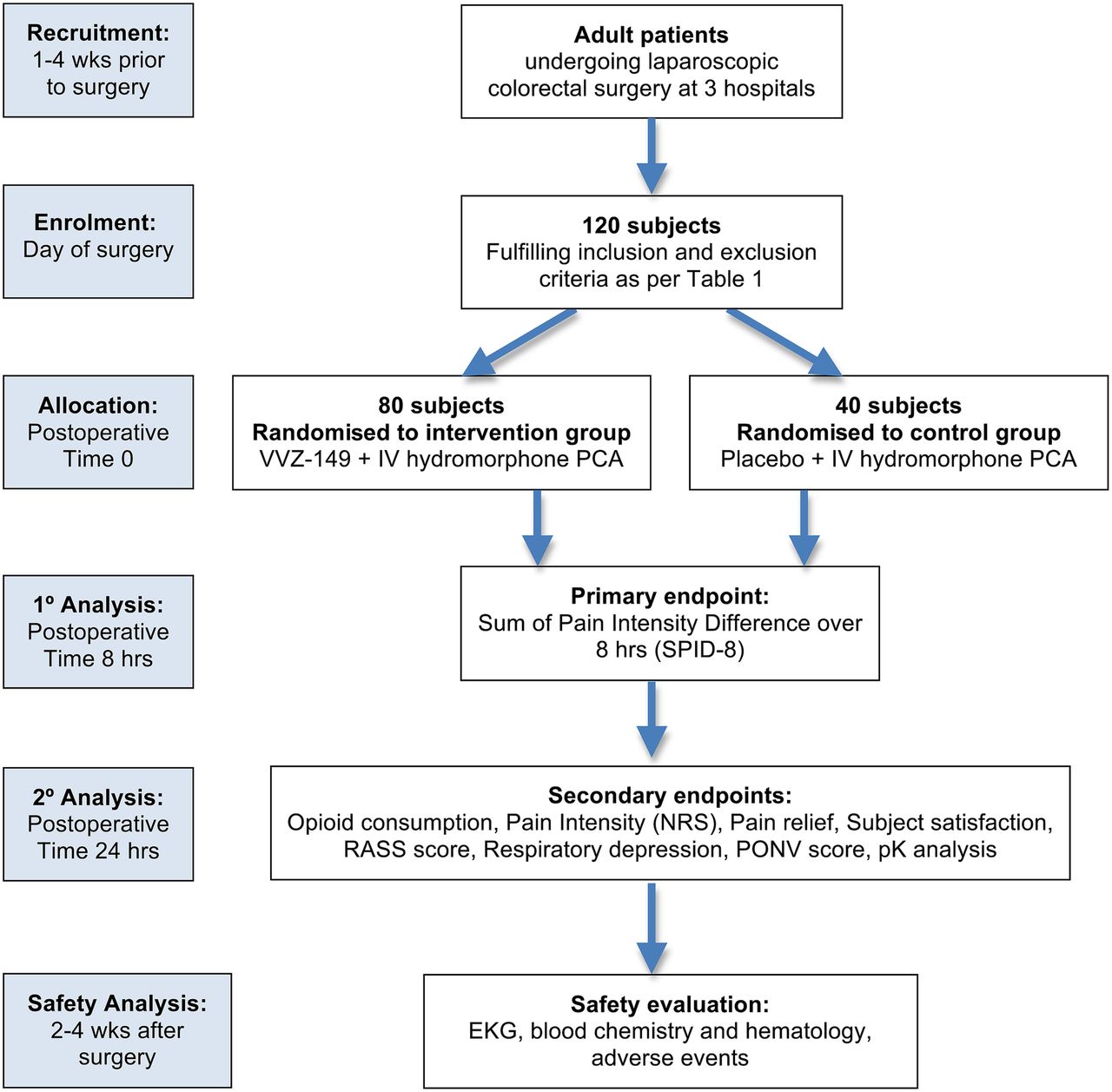
{kind=link}
CONSORT diagram for VVZ-149 study flow. IV, intravenous; NRS, Numerical Pain Rating Scale; PCA, patient-controlled analgesia; PONV, Post-operative Nausea and Vomiting scale; RASS, Richmond Agitation and Sedation Scale.
Setting and recruitment
Patients are being recruited from three academic medical centres: Brigham and Women's Hospital, Beth Israel Deaconess Medical Center, and Massachusetts General Hospital. All three hospitals are located in Boston, Massachusetts, and are teaching affiliates of Harvard Medical School. Enrolment is expected to conclude by December 2016.
Patients undergoing planned laparoscopic colorectal surgery are informed about the study at their local surgeon's office or at the facility's preoperative test centre, where they are asked if they would like to participate in the VVZ-149 trial. Prior to recruitment, members of the research team evaluate whether the patient is potentially an appropriate candidate for enrolment into the study based on diagnosis, age and the nature of the planned procedure. The physician or study representative introduces the study during the patient's planned visit to their surgeon or the preoperative test centre, or by telephone call for those patients who do not require prehospital evaluation within the facility. Each potential patient is given detailed information about the procedures involved with the study protocol as well as the Institutional Review Board (IRB)-approved consent form to review prior to agreeing to participate in the study. All patients are required to provide written informed consent when enrolling in the study.
Eligibility criteria
Inclusion and exclusion criteria for patients to participate in the study are listed in table 1. Inclusion criteria were developed to allow enrolment of adult patients age 70 or lower who are without high anaesthetic risk classification (American Society of Anesthesiologists risk class of I–III, ASA I–III) and who are scheduled to undergo laparoscopic colorectal surgery. Patients must have the ability to understand study procedures and communicate in English. To be enrolled in the study, a patient must have minimal pain intensity of ≥4 (on a 0–10 scale) at the time of their initial pain measurement after surgery in the PACU.
Inclusion and exclusion criteria for phase II VVZ-149 study
Patients may be excluded from participation in the study due to certain surgical factors, participant characteristics, anaesthetic factors or pharmacological considerations. Many of the exclusion factors were chosen to reduce or eliminate possible confounders that could affect study outcomes, like the use of neuraxial, regional or local anaesthesia or the use of certain analgesic drugs such as NSAIDs, anticonvulsants, ketamine, acetaminophen or herbal agents. Other exclusion factors were chosen to maintain safety of study participants, such as exclusion of patients who have prolonged QTc on their ECG, those with unstable or acute medical conditions, or those who have clinically important renal or hepatic impairment. To avoid the confounder of ongoing pain and opioid use, participants with a chronic pain diagnosis or those taking opioids on regular basis preoperatively were excluded. To ensure that treatment results are not affected by recent prior surgery or use of analgesics, we are excluding patients who have a cancer-related condition causing preoperative pain, those who are having a repeat operation within 30 days and those who are having emergency surgery.
Baseline assessments, treatment phase and post-treatment follow-up
Patients scheduled to undergo laparoscopic colorectal surgery are invited to participate in the study by the treating physician, research physicians and/or research staff. Potentially eligible and interested patients are then screened for eligibility by research staff at each of the sites. If a patient is eligible, the potential participant is asked to complete the informed consent process. A physician investigator reviews each potential patient's eligibility and preoperative test results (laboratory tests, ECG and response to questionnaires) to confirm the eligibility of the participant for enrolment into the study protocol.
On screening, questionnaires are administered to assess demographic and clinical factors, including medical and surgical history (Charlson-Katz questionnaire),23 medication history, vital signs, cognitive impairment (six-item Cognitive Screening questionnaire),24 alcohol/drug use (TICS questionnaire),25 ECG, and blood samples are obtained to assess baseline haematological, coagulation, renal, hepatic, thyroid and metabolic parameters. Patients are asked to complete self-report questionnaires to assess for anxiety, depression and pain catastrophising behaviours.26 ,27
Participants who meet eligibility criteria based on assessments carried out at screening check-in 2–4 hours prior to the scheduled surgery. At that time, any changes in medications are recorded and the eligibility of the participant is confirmed. Patients complete an ‘Expectations of Treatment’ questionnaire.28 Randomisation of eligible participants occurs during the time of surgery. After completion of surgery, participants are transferred to the PACU. There, it is confirmed that the participant continues to meet inclusion/exclusion criteria. An ECG is performed to ensure that there has been no significant increase in QTc interval that would otherwise render the patient ineligible. Pain intensity (NRS), level of sedation (Richmond Agitation and Sedation Scale, RASS), presence of postoperative nausea and vomiting (Post-operative Nausea and Vomiting scale, PONV), and level of respiratory depression are assessed. In patients who use postoperative IV PCA with opioids, a level of respiratory depression of <8 breaths per minute and an oxygen saturation level of <90% is considered clinically significant.29 On regaining a level of alertness (RASS score 1 or greater) and no more than 1 hour after emergence from anaesthesia, participants are asked to report a predose pain intensity score. Participants who indicate a pain score of at least 4 on the 11-point NRS are dosed with the study medication within 30 min of measuring the predose pain intensity score.
The experimental group receives a 1.8 mg/kg VVZ-149 intravenous infusion for 0.5 hour. This loading dose is followed by a maintenance dose of an intravenous VVZ-149 infusion of 1.3 mg/kg/hour for 7.5 hours.
A PCA pump containing hydromorphone (1 mg/mL) is connected to an intravenous infusion line and programmed to deliver 0.2–0.3 mg boluses of hydromorphone on demand by the participant, with a lockout time interval of 6 min. If adequate pain relief is not achieved with the IV PCA titration (pain score NRS≥6), a mandatory ‘rescue’ dose of 0.5 mg IV hydromorphone is administered as needed at a time interval of every 5 min to achieve a pain score of NRS≤5, but total number of rescue doses cannot be more than three times per hour.
Participants who no longer require IV PCA can be transitioned to oral hydromorphone (4 mg every 3–4 hours as needed) after 12 or more hours postdosing.
Once the treatment infusion has been initiated, participants are observed for 24 hours. Patients are placed on centrally monitored continuous pulse oximetry for up to 12 hours postdosing regardless of whether they are receiving VVZ-149 and while they are receiving IV PCA. In addition, respiratory depression assessments are carried out at predose and each postbaseline time point and prior to administering any rescue doses of hydromorphone. Participants complete the treatment phase of the study on postoperative day 1 after a 24-hour postdose assessment has been completed.
In participants who receive rescue dosing, the pain intensity (NRS) rating that is measured immediately before the dose is documented to replace the closest measurement scheduled to be recorded as part of the study protocol. If the number of rescue doses exceeds three times per hour, the participant is withdrawn from the study and allowed additional boluses of opioid and other analgesic treatments that are deemed clinically appropriate.
During the drug infusion phase (0–8 hours) and treatment assessment phase (8–24 hours), patients are periodically assessed for pain intensity at rest and with movement, pain relief, RASS,30 the incidence and clinical importance of PONV,31 opioid consumption, respiratory depression, adverse events and concomitant use of medications. Vital signs are recorded and ECGs are obtained at several time points. Blood samples for pharmacokinetic analysis are also obtained (predose, and then at 0.5, 1, 4, 8, 9, 12 and 24 hours after dosing). At 8 and 24 hours postdose, patient satisfaction with treatment is evaluated using the ‘Global Measure of Subject Satisfaction Scale’. At 24 hours postdosing, a blood sample to evaluate haematological, coagulation, renal, hepatic, thyroid and metabolic parameters is collected.
To assess the robustness of blinding, a ‘Perception of Treatment’ questionnaire is administered to study participants and to the study personnel who most closely observed the participant during the treatment phase of the protocol.32 Participant expectation of treatment and overall satisfaction with the study drug is evaluated to see if these parameters are associated with treatment outcomes.
Randomisation and blinding
In this double-blind study, the investigator, research assistants involved in obtaining data, the treating clinicians and the study participants are blinded to the treatment assignment of the study participants while the site pharmacist and study project manager remain unblinded.
After confirming the initial eligibility of participants, the randomisation is requested by the site project team during the time of surgery. The site pharmacist is (1) notified of the randomisation code generated via the electronic data capture system, (2) confirms which group the participant is assigned through the list of randomisation code and group provided to only the site pharmacist, (3) mixes VVZ-149 injections or placebo in a 500 mL saline IV bag after calculating the dosing volume according to the participant's weight, and (4) dispenses the IV bag to the nurse for the dosing.
The list of randomisation codes and group assignment lists are securely retained by the site pharmacist. The randomisation information of each participant can be disclosed to the investigator in an emergency situation only. If unblinding occurs, the study medication for the participant is discontinued and a written explanation of the event is prepared immediately.
Participants who are unblended will receive opioid analgesics as needed, per the standard of practice for postoperative pain management at the institution, and at the discretion of their clinician. On discontinuation of the study medication and completion of the study period at 24 hours after dosing, participants will be managed per the standard practice of the clinician following laparoscopic colorectal surgery.
Treatment administered and selection of doses in the study
The treatment consists of a loading dose of 1.8 mg/kg VVZ-149 injections administered by intravenous infusion for 0.5 hour, followed by a maintenance dose of 1.3 mg/kg/hour VVZ-149 injections for 7.5 hours. The placebo group receives the corresponding volume of placebo for the proscribed time of the study.
The estimated therapeutic range of targeted pooled plasma concentration has been determined to be 600–1900 ng/mL, which corresponds to 2–6 mg/kg in a 4-hour infusion as the Cmax of the dose in a phase 1 study (PT-VVZ149-01).21 Using Non-linear Mixed Effects Modeling (NONMEM) with individual pharmokinetic data from the phase 1 study, the expected plasma concentration and individual variability of an initial loading dose followed by a maintenance dose were simulated. The results suggest that a loading dose of 1.5 mg/kg of 0.5 hour followed by a maintenance dose of 1.1 mg/kg/hour for 7.5 hours will achieve a similar level of pooled plasma concentration at a steady state with Cmax of 5 mg/kg in a 4-hour IV infusion, a level within the range where VVZ-149 injections is expected to demonstrate its maximum efficacy. However, actual plasma concentration in a phase 1 study with elderly participants (PT-VVZ149-02) appeared to be about 20% less than the expected level by the simulation.21 Thus, the doses used for this protocol have been adjusted to 1.8 mg/kg of 0.5 hour for a loading dose followed by a maintenance dose of 1.3 mg/kg/hour for 7.5 hours to increase the plasma exposure level by about 20% of the level of maximally efficacious analgesic effect without adverse side effects.
Anaesthesia protocol
All participants have general anaesthesia without any regional or neuraxial anaesthesia, and participants do not receive ketamine or lidocaine >1 mg/kg. Only IV hydromorphone, fentanyl or morphine is administered as opioids during anaesthesia. The dose and type of IV opioid given intraoperatively is recorded as one of the variables that can affect treatment outcome. No more than the following doses are given intraoperatively: IV morphine 10 mg, IV fentanyl 500 µg total during entire anaesthetic and IV hydromorphone 2 mg. Within 1 hour of expected surgical completion, only IV fentanyl up to 200 µg is administered. Opioid doses are determined at the clinical discretion of the anaesthesiologist based on participants' surgical response and haemodynamic parameters while under anaesthesia. The amount of opioid administered intraoperatively will be converted to a total morphine equivalent dose and will be considered in the analysis of outcomes. No NSAID, gabapentin, pregabalin or acetaminophen is provided intraoperatively or during the postoperative study period. No more than 20 mL of 1% lidocaine anaesthetic wound infiltration may be given. At the discretion of the anaesthesiologist, participants may receive standard doses of intraoperative prophylactic or postoperative therapeutic antiemetics (dexamethasone ≤10 mg, haloperidol ≤1 mg, ondansetron ≤4 mg) and the use of these drugs is recorded.
After the completion of surgery, participants will be transferred to the PACU within 30 min of emergence from anaesthesia. In the PACU, the study protocol and the IV PCA pump will be ready to initiate as soon as possible once the anaesthesiologist delivers the participant to the PACU.
If there is a need to administer IV opioid from the time of emergence of the participant from general anaesthesia to the time they are transferred to the PACU and therefore before the study protocol is initiated, fentanyl up to 3 µg/kg may be administered incrementally as needed and the dose recorded.
Use of opioid PCA
An opioid PCA pump containing hydromorphone (0.5–1.0 mg/mL) is connected to an intravenous infusion line and programmed to deliver 0.2–0.3 mg boluses of hydromorphone on demand by the participant, with a lockout time interval of 6 min. If adequate pain relief is not achieved with the IV hydromorphone PCA (if pain score NRS is ≥6), a ‘rescue’ bolus of 0.5 mg/bolus IV hydromorphone may be administered as needed at a time interval of every 5 min to achieve a pain score of NRS ≤5, but the total number of rescue doses may not be given over three times per hour. In participants who receive rescue dosing, the pain intensity (NRS) rating that is measured immediately before the dose and the measurement is documented to replace the closest measurement scheduled to be recorded as part of the study protocol. If the number of rescue doses exceeds three times per hour, the participant is withdrawn from the study and allowed additional boluses and analgesic treatments that are deemed clinically appropriate. Participants who are withdrawn continue to be monitored as part of the study protocol and safety and efficacy data continue to be collected and recorded for future analysis.
Participants who no longer require IV PCA may be transitioned to oral hydromorphone (2–4 mg every 3–4 hours as needed) to replace IV PCA 12 hours or longer postdosing. The amount and frequency of oral hydromorphone used by the participant 12–24 hours postdosing is recorded, as well as any IV rescue dosing necessary during this time.
Outcomes
The primary outcome is the Sum of Pain Intensity Difference over 8 hours postdosing of the study drug (SPID-8).33 This is assessed using an 11-point NRS pain scores (0–10) measured up to 8 hours postdose. Pain intensity difference (PID) is calculated by subtracting each participant's baseline score of pain with movement minus postdosing pain scores so that positive differences indicated decreased pain. PID scores are evaluated using predose scores, and then at 0.25, 0.50, 1, 2, 4, 6 and 8 hours postdosing.
A number of secondary outcome measures are being analysed. Opioid consumption up to 24 hours postdosing of study drug at 0–2, 2–4, 6–8, 8–12, 12–16 and 16–24 hours postdosing of study drug is assessed while also evaluating the total number of PCA demands for opioid, the total number of rescue doses given and the time to the first rescue dose. Pain intensity at rest and with movement is assessed as recorded on the 11-point NRS. Categorical Pain Relief scores are assessed using a six-point scale (0: worse, 1: none, 2: a little relief, 3: some relief, 4: a lot of relief, 5: complete relief) up to 24 hours postdose. Using these data, Total Pain Relief (TOTPAR) over the 8-hour and 24-hour time points is calculated.34 Global Measurement of Subjects Satisfaction with the study medication is assessed using a five-point scale (extremely dissatisfied, 1: dissatisfied, 2: neutral, 3: satisfied, 4: extremely satisfied) at 8 and 24 hours postdose. Finally, side effects of the study medication are evaluated by analysing results of the RASS, PONV and by reviewing the respiratory depression assessments for up to 24 hours postdose.
As part of this efficacy and safety study, blood samples are collected from patients at various time points predosing and postdosing to perform pharmacokinetic evaluation of the study product. A pharmacokinetic to pharmacodynamic correlation assessment will eventually be performed.
Participants are assessed at one additional time point after discharge from the hospital, 14–30 days after surgery. At that time, vital signs are measured, an ECG is performed, and any adverse events or changes in medications are noted. A blood sample to evaluate haematological, coagulation, renal, hepatic, thyroid and metabolic parameters is collected.
Sample size calculation
The sample size for this study was determined based on the conjectures for the primary end point of SPID. Specifically, a minimum clinically important difference between group means of 5, with a maximum SD of 7, was conjectured. A statistical power of 90% with an overall α-level of 5% (4.5% for the final analysis and 0.5% for the interim analysis) was assumed in the calculations. In addition, a maximum attrition rate of 15% is incorporated. With all the above specifications, 120 participants with a 2:1 ratio will be enrolled into the study (n=80) and control (n=40) groups.
Statistical methods
Unless otherwise specified, standard descriptive statistics will be computed for all end points and other observed values. The standard descriptive statistics for continuous variables include: number of observations analysed, mean, SD, median, minimum and maximum. The standard descriptive statistics for categorical variables include: frequency distribution with the number and per cent of participants included in each category.
Calculation of percentage will use the denominator of the total number of participants in the particular group of analysis population used in the data display unless otherwise specified.
All observed data will be summarised and analysed. No imputations are planned for missing data.
Analysis of primary end point
SPID-8 will be summarised descriptively for each of the two groups. Groups will be compared using a t-test if the end point is approximately normally distributed, or using a Wilcoxon rank-sum test if the end point is markedly non-normally distributed.
Analysis of secondary end points
Opioid consumption, NRS, PID, RASS, respiratory depression assessment, Pain Relief, TOPAR, global assessment of satisfaction and intensity of PONV (number of instances and duration) will be summarised and analysed using the same methodology as described for the primary end point, SPID. The time to first rescue hydromorphone dose will be summarised with Kaplan-Meier estimates and groups will be compared with a log-rank test.
Analysis of potential confounders
Potential confounders of analgesic response include baseline levels of anxiety and depression, expectations of treatment, and catastrophising behaviour. For each potential confounder, we will construct an analysis of covariance model. To examine if the effect of treatment differs depending on any patient characteristics that precede treatment, we will assess statistical significance with a likelihood ratio test. We will evaluate baseline characteristics to assess if there are non-specific predictors of outcome that may have an impact on results in the active and placebo treatment groups. For an evaluation of the robustness of blinding (another potential confounder), we will use Fisher's exact test to compare treatment guesses between arms.
Safety analyses
Adverse events will be compared between groups using χ2 tests or Fisher's exact tests, as appropriate. The remaining safety end points will either be analysed using the same methodology as described for the primary end point, if continuous, or using χ2 tests or Fisher's exact tests, as appropriate, if categorical.
Pharmacokinetic analyses
The actual sampling time for each participant will be used in the PK analysis. Drug concentrations under lower limit of quantification (LLOQ), not applicable or not sampled will be recorded as <LLOQ, NA or ND, respectively. PK parameters will be calculated from the data from individual plasma concentration times of VVZ-149 Injections using non-compartmental methods. PK calculations will be performed using WinNonlin. The PK parameters of VVZ-149 injections will be calculated as per table 2.
Pharmacokinetic parameters collected in the VVZ-149 protocol
PK parameters will be summarised using descriptive statistics (eg, mean, SD, median, minimum, maximum and coefficient of variation). Plasma drug-concentration time curves will be expressed as linear or log/linear graphs for each participant, and the mean of the plasma drug-concentration time curve for each dose level will be used in the same manner.
Linearity and dose proportionality will be evaluated using linear regressions of AUClast, AUCinf, AUCτ,ss, Cmax and Cmax,ss to the dose and of log-transformed AUClast, AUCinf, AUCτ,ss, Cmax and Cmax,ss to the log-transformed dose. A graphical presentation of the linearity and dose proportionality will also be provided.
Interim analyses
When 60 participants complete the study, there will be a single blinded interim analysis planned. The main purpose of this blinded interim analysis will be futility (go/no go). The details of this interim analysis will be presented in the statistical analysis plan which will be finalised before the interim analysis.
Ethics and dissemination
The study is being conducted in accordance with Declaration of Helsinki standards and the Food and Drug Administration (FDA) regulations regarding Good Clinical Practice.
Informed consent
All participants will have been provided with information about the possible risks and benefits of participating in this clinical trial. After being given time and opportunity to ask questions, participants will be provided an informed consent form approved by the local IRB. A signed copy of the consent form will be maintained with the study records.
Participant confidentiality and dissemination of results
Trial participants will be given a unique participant identification number to maintain confidentiality, and no protected health information will be disseminated. Study results will be published in a scientific journal on study completion.
Discussion
Although significant strides have been made in treating postoperative pain, there continue to be patients in whom the level of pain creates dissatisfaction and impedes recovery. In colorectal surgery, the use of standard analgesics such as opioids is problematic due to the desire for a rapid return of bowel function and the tendency of these drugs to worsen or prolong ileus.35 NSAIDs can lead to bleeding problems, renal dysfunction or thrombotic events, which may have adverse effects on patient recovery.5 ,6 For those reasons, many surgeons have begun to adapt Enhanced Recovery After Surgery (ERAS) protocols. These protocols involve early mobilisation of patients after surgery, maintaining a euvolemic state by avoiding overhydration, and optimising pain management through multimodal techniques while minimising opioid use. Data on outcomes of ERAS protocols in patients undergoing colorectal surgery have been favourable.36
Various in vivo pharmacological studies with laboratory animals have consistently demonstrated dose-dependent analgesic or antiallodynic effects of VVZ-149 in rat models of postoperative pain, formalin-induced nociceptive/inflammatory pain and neuropathic pain. The analgesic effect of VVZ-149 has been shown to be due to a non-opioid mechanism and was not blocked by pretreatment with naloxone, an opioid-receptor antagonist, in the formalin model. VVZ-149 did not inhibit cyclooxygenase activity in an in vitro assay. These results suggest that the analgesic effect of VVZ-149 is neither based on opioid nor non-steroidal anti-inflammatory-based mechanisms. VVZ-149 is thus a non-opioid and non-NSAID analgesic candidate with morphine-comparable efficacy that may be able to provide analgesic benefits to patients.
Consistent with the implementation of ERAS protocols, we anticipate that the use of this novel non-opioid analgesic VVZ-149 will provide an alternate and novel method of reducing postoperative pain in patients undergoing laparoscopic colorectal surgery. We have developed a study protocol that evaluates the efficacy and safety of using VVZ-149 in this surgical population and our research will determine if the use of this drug leads to the improved management of postoperative pain. In addition to evaluating the primary end point of PID over time, we are assessing secondary outcomes such as opioid use, pain relief, sedation, nausea and overall levels of patient satisfaction with treatment. We have implemented recruitment strategies and engaged in collaborative relationships at three academic hospitals and we believe that we have a successful strategy that will improve our ability to recruit patients who fulfill inclusion and exclusion criteria into our trial. It is our hope that this study will provide high-quality data to show whether VVZ-149 is safe and efficacious for use as an analgesic for postoperative pain in this study population.
Acknowledgments
The authors thank all participating sites, clinicians and investigators for their support with the clinical study. Additionally, the authors thank the staff at KCRN, a contract research organisation, and especially Rachel Kim and Hugh Lee, for their oversight of this research.
References
Footnotes
Contributors SSN provided advice on designing and drafting the protocol, edited and approved the final version; prepared the protocol for manuscript submission and is the lead author for this paper; also manages the day-to-day running of this clinical trial. DJC provided advice on parts of the protocol relating to surgical postoperative management. He is the clinical lead for the project at Brigham and Women's Hospital (BWH). XB provided advice on parts of the protocol relating to anaesthetic and surgical postoperative pain management. He is the clinical lead for the project at Massachusetts General Hospital (MGH). NZ provided advice on parts of the protocol relating to anaesthetic and surgical postoperative management. She is the clinical lead for the project at Beth Israel Deaconess Medical Center (BIDMC). JLZ provided advice on parts of the protocol relating to anaesthetic intraoperative management. He is involved in executing the protocol at BWH. YZ provided advice on parts of the protocol relating to surgical postoperative pain management. He is involved in executing the protocol at MGH. MJY provided advice on parts of the protocol relating to surgical postoperative pain management. He is involved in executing the protocol at BIDMC. JL and JS provided input regarding parts of the protocol regarding participant enrolment, data collection and coordination with pharmacy and laboratory service at BWH. KE provided input regarding parts of the protocol regarding participant enrolment, data collection and coordination with pharmacy and laboratory service at MGH. CPH, RM and JWC provided input regarding parts of the protocol related to postoperative data collection and PK sampling at BWH. BT provided input regarding parts of the protocol related to postoperative data collection and PK sampling at MGH. SC conceived of and planned the trial, provided basic science input, and approved the final version. DHL conceived of and planned the trial, drafted the protocol and approved the final version. All authors contributed to development of the research protocol, manuscript writing and editing of this protocol paper. All of the authors read and approved the final version of the protocol and of the manuscript submission.
Funding Funding to conduct this study is being provided by Vivozon, a privately held company based in South Korea, supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health and Welfare, Republic of Korea (grant number: HI15C2006).
Competing interests The following authors confirm that research grant funding has been assigned by the study sponsor, Vivozon, to Brigham and Women's Hospital, Massachusetts General Hospital, and Beth Israel Deaconess Medical Center, which are the employing institutions of authors SSN, DJC, XB, JLZ, YZ, MJY, JL, JS, KE, CPH, RM, JWC and BT. In addition, SSN received consulting fees from Vivozon. SC and DHL are employees of Vivozon Company, the sponsor of this study, and have received funding from the South Korean Ministry of Health and Welfare for R&D support for pharmaceutical development of the product that is described in this manuscript.
Ethics approval Ethical approval of the study protocol has been obtained from Institutional Review Boards in July 2015 at the participating institutions. Two of the three participating clinical sites (BWH and MGH) are under the auspices of a single IRB, the Partners Human Research Committee, and the third site (BIDMC) is under review by its own IRB, the Committee on Clinical Investigations.
Provenance and peer review Not commissioned; externally peer reviewed.