Article Text

Abstract
Introduction Despite the observed and theoretical advantages of shared decision-making in a range of clinical contexts, including contraceptive care, there remains a paucity of evidence on how to facilitate its adoption. This paper describes the protocol for a study to assess the comparative effectiveness of patient-targeted and provider-targeted interventions for facilitating shared decision-making about contraceptive methods.
Methods and analysis We will conduct a 2×2 factorial cluster randomised controlled trial with four arms: (1) video+prompt card, (2) decision aids+training, (3) video+prompt card and decision aids+training and (4) usual care. The clusters will be clinics in USA that deliver contraceptive care. The participants will be people who have completed a healthcare visit at a participating clinic, were assigned female sex at birth, are aged 15–49 years, are able to read and write English or Spanish and have not previously participated in the study. The primary outcome will be shared decision-making about contraceptive methods. Secondary outcomes will be the occurrence of a conversation about contraception in the healthcare visit, satisfaction with the conversation about contraception, intended contraceptive method(s), intention to use a highly effective method, values concordance of the intended method(s), decision regret, contraceptive method(s) used, use of a highly effective method, use of the intended method(s), adherence, satisfaction with the method(s) used, unintended pregnancy and unwelcome pregnancy. We will collect study data via longitudinal patient surveys administered immediately after the healthcare visit, four weeks later and six months later.
Ethics and dissemination We will disseminate results via presentations at scientific and professional conferences, papers published in peer-reviewed, open-access journals and scientific and lay reports. We will also make an anonymised copy of the final participant-level dataset available to others for research purposes.
Trial registration number ClinicalTrials.gov Identifier: NCT02759939.
- gynaecology
- primary care
- public health
- shared decision-making
- contraception
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Background and rationale
Shared decision-making is the process of healthcare providers and patients making health decisions together. While a number of conceptual definitions have been offered,1 the most common considers shared decision-making a process in which providers and patients exchange information, deliberate about available options together and come to agreement on the option to implement.2 Shared decision-making is conceptually distinct from paternalistic decision-making, which assumes that the patient’s preferences have no place in the decision-making process and from informed decision-making, which assumes that the provider’s only legitimate role in the decision-making process is information provision.2 3
Proponents of shared decision-making have described it as an ‘ethical imperative’4 and the ‘pinnacle of patient-centred care’.5 There is also a growing body of evidence of its generally positive impact on patient affective-cognitive, behavioural and health outcomes in a range of clinical contexts.6 In contraceptive care, shared decision-making may represent a strategy for promoting health and well-being while safeguarding patient autonomy, the importance of which is emphasised in key practice guidelines.7 8 For example, shared decision-making about contraceptive methods may facilitate the prevention of unintended or unwelcome pregnancies by increasing the likelihood that people choose methods aligned with their individual preferences and values, are satisfied with the methods they adopt and use them in a way that takes full advantage of their potential contraceptive and other benefits. The anticipated gains from shared decision-making about contraceptive methods may be particularly pronounced for members of minority racial and ethnic groups and people of low socioeconomic status, who experience significant disparities in contraceptive care and outcomes.9
Despite the numerous observed and theoretical advantages of shared decision-making, there remains a paucity of evidence on how to facilitate its adoption. A recent systematic review of interventions for improving the adoption of shared decision-making by healthcare providers found that, while interventions targeting patients alone (eg, question prompt lists) or providers alone (eg, audit and feedback) had some—if modest—positive effects on the adoption of shared decision-making, effects were generally greater when interventions targeted both groups.10 However, based on the small number and poor quality of available studies, reviewers were tentative in their conclusions and advocated for further research to respond to this important knowledge gap. Answering this call, this paper describes the protocol for a study that will assess the comparative effectiveness of patient-targeted and provider-targeted interventions for facilitating shared decision-making about contraceptive methods in the healthcare visit.
Objectives
The first objective of this study is to evaluate the effect of a patient-targeted intervention (a video+prompt card that encourages patients to ask three specific questions in the healthcare visit) and a provider-targeted intervention (decision aids+training for providers in their use; see Interventions), on shared decision-making about contraceptive methods in the healthcare visit. The second objective is to evaluate the effect of these interventions on the occurrence of a conversation about contraception in the healthcare visit, satisfaction with the conversation about contraception, intended contraceptive method(s), intention to use a highly effective method, values concordance of the intended method(s), decision regret, contraceptive method(s) used, use of a highly effective method, use of the intended method(s), adherence, satisfaction with the method(s) used, unintended pregnancy and unwelcome pregnancy (see Outcomes and measures). The third objective is to evaluate the feasibility of the interventions (operationalised as rates of patient exposure to the interventions) and their acceptability to patientsi.
Research questions
There are four research questions pertaining to the first study objective: (1) does implementing the video+prompt card increase the rate of shared decision-making about contraceptive methods compared to usual care? (2) does implementing the decision aids+training increase the rate of shared decision-making about contraceptive methods compared to usual care? (3) does implementing the video+prompt card and the decision aids+training result in greater increases in the rate of shared decision-making about contraceptive methods compared to usual care than implementing either of the interventions alone? and (4) what patient characteristics and other factors modify the effect of implementing the interventions on the rate of shared decision-making about contraceptive methods? Research questions pertaining to the second and third study objectives as well as study hypotheses are available in the online supplementary file.
Supplementary file 1
Methods
Trial design
We will conduct a 2×2 factorial cluster randomised controlled trial with four arms: (1) video+prompt card, (2) decision aids+training, (3) video+prompt card and decision aids+training and (4) usual care. This design will enable us to assess the effect of the two interventions, both alone and together, as compared with usual care. The clusters will be clinics (see Study setting) and participants will be a subset of patients attending these clinics (see Participants). While a cluster randomised controlled trial requires more participants than would a trial with a different approach to randomisation, we consider a cluster design most robust to prevent the contamination possible if the unit of randomisation were to be patients or providers.11
Study setting
The trial will take place in 16 clinics in the Northeast United States, permitting the recommended minimum of four clusters per trial arm to minimise potential confounding from cluster effects.11 Participating clinics may be primary care and/or reproductive healthcare clinics but must deliver contraceptive care, which we define as providing contraceptive methods on site, prescribing or referring people for contraceptive methods and/or counselling people about contraceptive methods. Participating clinics must also have sufficient patient flow that staff are confident to be able to facilitate 10 eligible patients, on average, completing a postvisit survey each week. Within each clinic, contraceptive care may be provided by any person with relevant training (eg, a physician, nurse, physician assistant, nurse practitioner, midwife, non-medically licensed counsellor). Participating clinics may operate in hospital or community settings, be located in rural or urban areas, be publicly or privately funded and be for-profit or not-for-profit. Participating clinics may deliver services in more than one geographic location but, to minimise contamination, may not employ someone who delivers contraceptive care in another participating clinic and may not employ a study investigator. Participating clinics will receive monetary compensation for the time expended by clinic staff on data collection and other administrative activities related to their role in the study. A list of participating clinics may be requested.
Interventions
Video+prompt card (patient-targeted intervention)
We developed a brief video intended to be viewed by patients in the clinic immediately before the healthcare visit. The video was designed to enhance patients’ motivation, skills and self-efficacy to ask their providers three questions in the healthcare visit: (1) what are my options? (2) Wwhat are the possible pros and cons of those options? and (3) How likely are each of those pros and cons to happen to me? Earlier iterations of these questionsii increased shared decision-making when used by unannounced standardised patients in a primary care setting in Australia12 and were the focus of the AskShareKnow programme, a multipronged intervention that was feasible to implement in a reproductive and sexual healthcare setting in Australia and acceptable to patients.13 The video was adapted from a 4-min video that comprised one component of the AskShareKnow programme.13 Our adaptation features a patient sharing a personal account of her experiences of healthcare, communicating the benefits of asking questions, normalising challenges associated with asking questions and encouraging people to ask the three target questions. The format and content of the adaptation responded to patient and stakeholder perspectives solicited via discussions among the research team (which includes patient partners and stakeholder representatives), focus groups with patients and consultation with the patient featured in the adaptation, who has since joined our research team. We developed versions of the video in English and Spanish, each with and without on-screen captions. We will supply clinics with tablet computers programmed to view the videos and headphones.
We also developed a small prompt card intended to be distributed to patients who view the video. The card was designed to remind patients of the three questions presented in the video. The prompt card was adapted from a refrigerator magnet that comprised another component of the AskShareKnow programme.13 Again, we developed versions of the prompt card in English and Spanish. We will supply clinics with the prompt cards and display stands.
Decision aids+training (provider-targeted intervention)
We developed seven one-page decision aids on contraceptive methods intended to be used by providers with patients during the healthcare encounter. There are decision aids on long-acting reversible contraceptive methods, short-acting reversible methods, barrier methods, natural methods, permanent methods and emergency methods as well a decision aid that provides an overview of these six categories of contraceptive methods. The decision aids were designed to help providers facilitate shared decision-making about contraceptive methods in the healthcare visit. The format of the decision aids was adapted from that used in Option Grid decision aids for clinical encounters, which have been found to increase shared decision-making in osteoarthritis care14 and to be acceptable to physicians.15 We engaged patients and stakeholders in developing the decision aids, including via a survey of patients and contraceptive care providers,16 patient focus groups and review of decision aid iterations by patient partners and stakeholder representatives. We developed versions of the decisions aids in English and Spanish. We will supply clinics with tear-pads of the decision aids and display stands.
We also developed a 5-min training video and accompanying written guidance intended to be viewed by providers before beginning to use the decision aids (and as frequently as desired thereafter). The training video and frequently asked questions were designed to enhance providers’ motivation, skills and self-efficacy to use the decision aids to facilitate shared decision-making in the healthcare visit. The content of the training video was informed by the Theoretical Domains Framework,17 18 which was devised to guide implementation research that involves healthcare provider behaviour change. The video features an Obstetrician-Gynaecologist, Nurse Practitioner and patient representative explaining how decision aids can support the delivery of quality healthcare and providing guidance on using the decision aids with patients. The written guidance reinforces and elaborates on training video content. We will host the training video and frequently asked questions on the study website. One or more clinic representatives will be charged with encouraging and enabling relevant healthcare providers in the clinic to review the training video and written guidance.
Implementation of interventions
The strategies we developed to support adoption of the interventions purposefully omitted elements difficult to scale (eg, face-to-face training by intervention developers, periodic feedback on rates of patient exposure to the interventions) to maximise the ecological validity of study findings. For the video and prompt card, we developed a set of presentation slides for clinic staff that provides guidance on their objectives and implementation. The slide deck will be hosted on the study website, along with a preview of the video and prompt card. One or more clinic representatives will be charged with encouraging and enabling others in the clinic to review the slide deck. For the decision aids and training, we also developed a set of presentation slides for clinic staff that provides guidance on their objectives and implementation. The slide deck will be hosted on the study website. Again, one or more clinic representatives will be charged with encouraging and enabling others in the clinic to review the slide deck. To further approximate real-world conditions, we will not prevent clinics from implementing concomitant interventions or care during the study.
Data collection
We will collect study data via longitudinal patient surveys administered immediately after the index healthcare visit (T1), four weeks (ie, 28 days) after the index healthcare visit (T2) and six months (ie, 182 days) after the index healthcare visit (T3) (see figure 1). Participants may elect to complete surveys in English or Spanish. The T1 survey will be administered in clinics using tablet computers and the Qualtrics online survey platform.19 At the end of the T1 survey, we will invite a subset of participants (see Participants) to give permission to be recontacted for the T2 and T3 surveys. Participants aged 20 years and older may elect to complete these surveys online (with email correspondence) or on paper (with postal mail correspondence). Participants aged under 20 years may only elect to complete these surveys online (with email correspondence) to safeguard their privacy. A unique password will be used to link participant responses across surveys. Copies of the surveys may be requested.

Data collection schematic
We have devised several strategies to maximise data quality. We chose to collect data via surveys rather than interviews given the vulnerability of the topic area to social desirability bias.20 For the T1 survey, which will be administered in clinics and collect data on participants’ care experiences and evaluations, we elected to use an online survey completed via tablet computer to ensure no handling of surveys by clinic staff and thus reinforce the confidentiality of responses. The study information sheet also reassures participants that no healthcare providers from participating clinics will have access to identified participant-level data. For all surveys, we used programmed or instructional skips to ensure that participants will be asked only relevant questions, thereby minimising survey fatigue. In the online surveys, we also implemented additional data quality strategies, such as pop-up messages that notify participants of missed questions and invite them to respond prior to proceeding to the next page.
We will also administer the T1, T2 and T3 surveys to a group of patients who attend participating clinics before we begin the trial. This ‘pilot’ data collection will replicate the trial data collection and thus, allow clinic staff time to become proficient in data collection processes. A further advantage of this pilot data collection is that we can assess equivalence between clinics in the usual rate of shared decision-making about contraceptive methods, adopt stratified assignment of clinics to trial arms if warranted (see Assignment) and statistically adjust for the usual rate of shared decision-making in relevant data analyses.
Assignment
Each clinic will be assigned to one of the four trial arms using permuted-block randomisation with an equal allocation ratio to achieve balance in the number of clinics per trial arm. Should we observe non-equivalence between clinics in the rate of shared decision-making about contraceptive methods during the pilot data collection period, we will adopt stratified permuted-block randomisation. Specifically, we will rank clinics according to the rate of shared decision-making and construct four strata based on this ranking, with one clinic assigned to each trial arm in each stratum. The study statistician (TDT) and statistical analyst (ZL) will generate the allocation sequence and assign clinics to trial arms. Due to the study design, it is not feasible to blind clinic staff, study participants or researchers to the trial arm to which each clinic has been assigned.
Participants
People who have completed a healthcare visit at a participating clinic, were assigned female sex at birth, are aged 15–49 years, are able to read and write English or Spanish and have not previously participated in the study will be eligible for the T1 survey. People who have not completed a visit at a participating clinic during the study period (including a patient’s parent or a person acting as a patient’s legal proxy), were not assigned female sex at birth, are aged under 15 or over 49 years, are unable to read and write English or Spanish or have previously participated in the study will be ineligible for the T1 survey. To enable us to answer study research questions without unnecessary participant burden, additional eligibility criteria will be imposed for the T2 and T3 surveys. People who completed the T1 survey, experienced a contraceptive conversation in the healthcare visit and intended to use one or more contraceptive methods following the visit (see Outcomes and measures) will be eligible for the T2 and T3 surveys. People are not required to have completed the T2 survey to be eligible for the T3 survey.
Outcomes and measures
We consulted with patients and other stakeholders in the process of selecting study outcomes. We prioritised the inclusion of patient-centred outcomes, including participants’ perceptions of the values concordance of their intended contraceptive method(s), decision regret pertaining to their intended contraceptive method(s) and satisfaction with the contraceptive method(s) used. To maximise the utility of study results for different audiences and purposes, we also elected to include a small number of more conventional, public health-oriented outcomes such as use of a highly effective contraceptive method.21 All primary and secondary study outcomes are presented in table 1 and elaborated below.
Outcomes and timing of measurement
To maximise data quality, we selected measures for the primary and secondary outcomes based on their brevity, readability, availability in English and Spanish, psychometric properties, prior use in other studies or population-level surveys and use of patient-centred language and tone. Where we identified a suitable measure in English that was not available in Spanish, we arranged for it to be translated. Where we could not identify a suitable measure, we developed or adapted one in English and arranged for it to be translated.
Primary outcome
Shared decision-making about contraceptive methods
We will measure shared decision-making about contraceptive methods in the healthcare visit using the three-item CollaboRATE.22 23 CollaboRATE was developed in consultation with end users23 and assesses people’s perceptions of the extent to which their healthcare provider(s) shared information, elicited their preferences and ensured their preferences were integrated as decisions were made. We will use the version of CollaboRATE with a five-point response scale and adopt the binary scoring approach.22 When used in this way in an experimental validation study, CollaboRATE demonstrated concurrent validity via a strong, positive correlation with the 9-Item Shared Decision Making Questionnaire24 and a moderate, positive correlation with the five-item Doctor Facilitation subscale of the Perceived Involvement in Care Scale.22 25 CollaboRATE also demonstrated discriminative validity, intrarater reliability and sensitivity to change.22 Because the CollaboRATE items are not condition-specific, we will use a customised opening statement to orientate participants to the conversation about contraception they experienced in the healthcare visit.iii
Secondary outcomes
Conversation about contraception
We will measure whether participants experienced a conversation about contraception in the healthcare visit using a self-developed item. Due to divergent perspectives on the circumstances under which a contraceptive conversation is indicated, we will report this outcome among (1) all participants, (2) all participants except those not at risk of unintended pregnancy (see Other data for definition and measurement) and (3) all participants except those not at risk of unintended pregnancy and those who reported that they did not want or need to talk about contraception.
Satisfaction with conversation about contraception
We will measure participants’ satisfaction with the conversation about contraception in the healthcare visit using an item adapted from Weisman et al.26
Intended contraceptive method(s)
We will measure what, if any, contraceptive method(s) participants intend to use in the next 4 weeks using a self-developed checklist of 20 methods. The checklist specifies common and brand names for some methods, encourages participants to review explanatory information about the methods if unsure and allows participants to select multiple methods or select ‘none of these’. We will report this outcome among (1) all participants, (2) all participants except those not at risk of unintended pregnancy (see Other data for definition and measurement) and (3) all participants except those not at risk of unintended pregnancy and those who reported that they did not want or need to use a birth control method.
Intention to use a highly effective contraceptive method
We will use data on participants’ intended contraceptive method(s) to derive a variable that represents whether they intend to use at least one highly effective contraceptive method in the next 4 weeks. We consider highly effective contraceptive methods to comprise the copper intrauterine device (IUD), the hormonal IUD, the contraceptive implant, female sterilisation and male sterilisation, all of which have a typical-use unintended pregnancy rate of <1% in the first year of use.27 We will report this outcome among (1) all participants, (2) all participants except those not at risk of unintended pregnancy (see Other data for definition and measurement) and (3) all participants except those not at risk of unintended pregnancy and those who reported that they did not want or need to use a birth control method.
Values concordance of intended contraceptive method(s)
We will measure participants’ perceptions of the values concordance of their intended contraceptive method(s) (ie, the degree of alignment between the method(s) and their individual values and preferences) using the self-developed, single-item Measure of Alignment of Choices (MATCH). MATCH contains an opening statement that orients the participant to the contraceptive method(s) of interest and then asks either ‘How confident are you that this method is right for you?’ or ‘How confident are you that these methods are right for you?’ When we administer this measure at T2 and T3, we will remind participants of their nominated intended contraceptive method(s).
Decision regret about intended contraceptive method(s)
We will measure participants’ feelings of decision regret about the contraceptive method(s) they intended to use using an adaptation of the five-item Decision Regret Scale.28 29 When we administer this measure at T2 and T3, we will remind participants of their nominated intended contraceptive method(s).
Contraceptive method(s) used
We will measure what, if any, contraceptive method(s) participants used in the last 4 weeks using an adaptation of the checklist used to assess intended contraceptive method(s).
Use of a highly effective contraceptive method
We will use data on the contraceptive method(s) participants used to derive a variable that represents whether they used at least one highly effective contraceptive method in the last 4 weeks.
Use of intended contraceptive method(s)
We will use data on participants’ intended contraceptive method(s) and the contraceptive method(s) they used to derive a variable that represents whether participants used their intended contraceptive method(s) in the last 4 weeks.iv
Adherence to contraceptive method(s) used
We will measure participants’ adherence to the contraceptive method(s) they used in the last 4 weeks using the self-developed, 21-item Adherence to Birth Control (ABC) measure.
Satisfaction with contraceptive method(s) used
We will measure participants’ satisfaction with the contraceptive method(s) they used in the last 4 weeks using a self-developed item. We will use a slightly modified version of this item to measure satisfaction among participants who reported that they used none of the listed contraceptive methods in the last 4 weeks.
Unintended pregnancy (pregnancy timing preferences)
We will measure participants’ experience of one or more unintended pregnancies, as defined by their pregnancy timing preferences, after the healthcare visit. For each pregnancy experienced after the healthcare visit, we will measure the pregnancy timing preferences participants’ held immediately before conception using an item adapted from the Pregnancy Risk Assessment Monitoring System (PRAMS) Phase 7 questionnaire.30 We will classify participants either as having experienced unintended pregnancy or not having experienced unintended pregnancy based on their responses.
Unintended pregnancy (pregnancy seeking)
We will measure participants’ experience of one or more unintended pregnancies, as defined by their pregnancy seeking, after the healthcare visit. For each pregnancy experienced after the healthcare visit, we will measure participants’ pregnancy seeking immediately before conception using an item adapted from Kavanaugh and Schwarz.31 We will classify participants either as having experienced unintended pregnancy or not having experienced unintended pregnancy based on their responses.
Unwelcome pregnancy
We will measure participants’ experience of one or more unwelcome pregnancies after the healthcare visit. For each pregnancy experienced after the healthcare visit, we will measure participants’ feelings on learning about the pregnancy using an item adapted from the PRAMS Phase 7 questionnaire.30 We will classify participants either as having experience unwelcome pregnancy or not having experienced unwelcome pregnancy based on their responses.
Process outcomes
Intervention exposure and question asking
We will measure participants’ exposure to the video, receipt of the prompt card and use of the three questions in the healthcare visit using five items adapted from Shepherd et al,13 administered at T1. We will measure participants’ exposure to one or more of the decision aids (including nature and timing of exposure) using a self-developed item administered at T1.
Acceptability of interventions
We will measure the acceptability of the video, prompt card and decision aid(s) to participants using three self-developed items administered at T1.
Exposure to concomitant interventions
We will measure exposure to other information on contraception on the day of the healthcare visit using a self-developed item administered at T1.
Other data
Visit date
The T1 survey will automatically record the date and time of survey completion (and thus, date of the healthcare visit) for each participant. We will use these data to distinguish between pilot and trial participants.
Clinic
We will measure the clinic in which each participant had their healthcare visit (and thus, trial arm) using a self-developed item measure administered at T1. The T1 survey will also automatically record the Internet Protocol address of the tablet computer on which the T1 survey is completed. We will use these data to review the accuracy of patient-reported data on clinic, managing inconsistencies on a case-by-case basis.
Previous contraceptive method(s)
We will measure what, if any, contraceptive method(s) participants used in the 4 weeks before the healthcare visit using an adaptation of the checklist used to assess intended contraceptive method(s).
Pregnancies
We will measure the number of pregnancies participants experienced after the healthcare visit using a self-developed item administered at T3. When we administer this item, we will remind participants of the date of their healthcare visit.
Explanatory questions and risk of unintended pregnancy
At T1, we will ask participants who did not experience a conversation about contraception to report one or more reasons for this from a self-developed list. At T1, we will also ask participants who do not intend to use any of the contraceptive methods in the checklist to report one or more reasons for this from a self-developed list. We will use the self-reported data participants provide in response to these questions to identify those not at risk of unintended pregnancy at the time of the healthcare visit. We will consider participants to be not at risk of unintended pregnancy if they report that they are pregnant, are trying to get pregnant, do not have ovaries or a uterus, have entered menopause, are infertile or do not plan to have vaginal sex with a person that produces sperm.
At T2 and T3, we will ask participants who had not used their intended contraceptive method(s) in the last 4 weeks to report the reason(s) for this in an open-ended question. When administering the ABC measure at T2 and T3, we will provide a free text box after the adherence question(s) for each method to enable participants to elaborate on their response(s). We will also provide a free text box at the end of the T2 and T3 surveys to enable participants to share any other information they choose.
Participant and visit characteristics
We will measure several participant and visit characteristics at T1. We will measure participants’ age, ability to read and write English or Spanish, sex assigned at birth,32 visit completion, previous study participation and clinic (see above) to confirm study eligibility. We will also measure participants’ current gender identity,32 race, ethnicity and educational attainment,33 health insurance coverage34 and health literacy.35–39 We will measure participants’ reproductive history (ie, number of pregnancies, births, abortions and miscarriages) using four self-developed items. We will also document the language in which each participant completed the survey.
Recruitment and retention
Prospective participants will be made aware of the opportunity to take part in the study on the day of their healthcare visit via study posters and information sheets displayed in participating clinics and/or through communication with clinic staff. We have adopted several strategies for achieving adequate participant enrolment and maximising participant retention, informed by both reviews of empirical evidence40 41 and patient and stakeholder perspectives. To help achieve adequate participant enrolment, we developed engaging recruitment materials that use a study name and branding designed with patient input. We adopted inclusive eligibility criteria (see Participants) and elected to allow participation without a commitment to complete all three surveys. We will also compensate participants with a $10 gift card for completing the T1 survey.
To maximise retention of participants enrolled in the study and minimise nonresponse error, we adopted both online and paper completion modes for the T2 and T3 surveys for participants aged 20 years and older (see Data collection). We will provide people who elect to complete the surveys on paper with an addressed, reply-paid envelope to facilitate survey return. We elected to administer the sending and receiving of T2 and T3 surveys from the research institution, thereby removing the responsibility from clinic staff. We have also developed survey invitation and reminder schedules customised to the survey mode. For the online completion mode, we will send two daily email reminders to participants who have not completed the T2 or T3 survey and will also send an email prompt 1 week in advance of the T2 survey invitation to encourage completion on the target date (see figure 2). For the paper completion mode, we will send a similar mail prompt 1 week in advance of the T2 and T3 survey invitations, but will not send daily mail reminders due to anticipated variation in mail delivery times and the potential for such reminders to arrive simultaneously or overlap (see figure 2). Again, we will compensate participants with a $10 gift card for completing each of the T2 and T3 surveys.
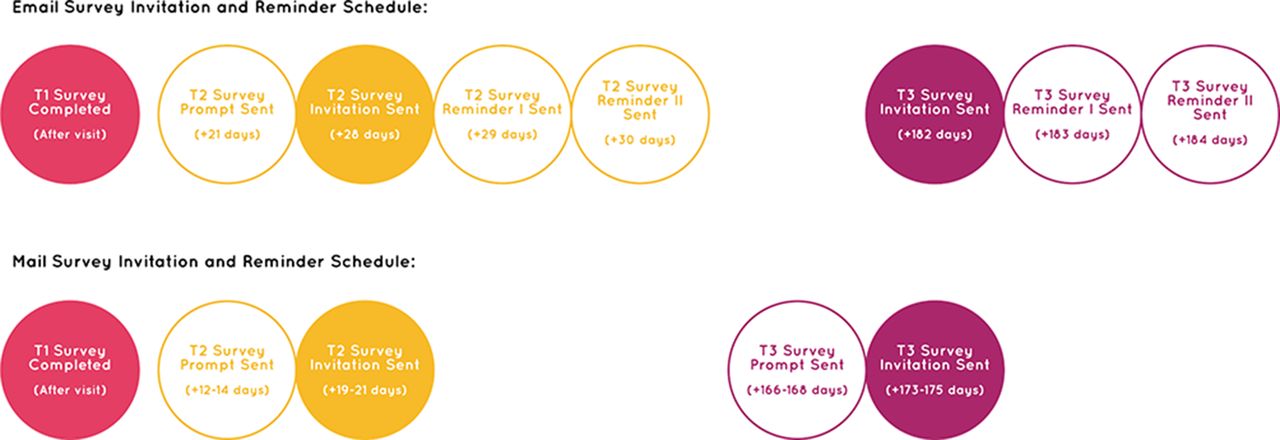
Survey invitation and reminder schedule.
Sample size and power
Sample size
Each of the 16 participating clinics will be expected to facilitate 10 eligible patients completing the T1 survey per week on average. Thus, we estimate that 1040 participants per trial arm will complete the T1 survey during the trial period. We estimate that 728 participants per trial arm (70%) will experience a conversation about contraception and thus comprise the sample for the primary outcome analysis. We estimate that 70% of the participants who experience a contraceptive conversation will be eligible for and agree to be invited to complete the T2 and T3 surveys. Of these, we estimate that 80% will complete the T2 survey and that 70% will complete the T3 survey (see figure 3).

{kind=link}
{kind=link}
{kind=link}
Estimated sample sizes per trial arm. *Estimates of the total number of patients eligible for the study will be provided by clinics based on routinely collected data. ^Sample for primary outcome analyses.
Additionally, we estimate that, in each clinic, 130 pilot participants will complete the T1 survey, 91 pilot participants will experience a conversation about contraception and 51 and 45 pilot participants will complete the T2 and T3 surveys, respectively.
Power
The study was powered to answer Research Questions 1, 2 and 3 (see Research questions). As outlined above, we estimate a sample size of 728 participants per trial arm for the primary outcome analysis (see figure 3). We base our detectable difference calculations on tests comparing the proportion of participants who experience shared decision-making about contraceptive methods (the primary outcome) between trial arms, assuming a proportion of 66% in the usual care arm.42 Given the fixed number of clusters per trial arm and the estimated sample size, we determine a detectable increase of 16% (from 66% to 82%) in the proportion of participants who experience shared decision-making, based on a z-test comparing two proportions with clustered data, with an estimated intracluster correlation coefficient of 0.03,43 a two-sided significance level of 5% and a power of 80%. If we apply a Bonferroni correction for the three possible comparisons versus the usual care arm to retain a nominal family-wise significance level of 5%, we can detect an increase of 18.1% in the proportion of participants who experience shared decision-making. These calculations show that we will have a sufficient sample size to detect meaningful changes in shared decision-making. All calculations were done using the PASS 2008 sample size package44 based on standard methods.45
Data management and analysis
Data management
The surveys will collect direct and indirect participant identifiers. To minimise risks to participant privacy, only three people from the study team (the principal investigator, the project manager and the data manager) will be allowed to access highly identifying participant-level data and files. The people in these roles will be responsible for sending and receiving T2 and T3 surveys and associated correspondence; entering, cleaning and recoding data and securely storing and transferring identified participant-level data and files. They will also be responsible for deleting or destroying highly identifying information (ie, participant names, email addresses and mailing addresses) from all documents and files on the completion of the study and otherwise anonymising data prior to sharing it. Our anonymisation process will take into account 28 direct and indirect patient identifiers.46 To maximise the utility of study data, we will store both identified data (excluding highly identifying information) and anonymised data indefinitely. A comprehensive data management plan may be requested.
Data analysis
Analysis plan
A data analysis plan is available in the online supplementary file 1.
Protocol non-adherence
While we will report rates of protocol non-adherence (derived from patient reports of exposure to the intended intervention(s)), the analyses for Research Questions 1, 2 and 3 will be conducted by intention to treat. Patient exposure to the interventions may comprise one factor included in the analyses for Research Question 4.
Treatment of missing data
We will report rates of and reasons for missing data, whether due to unanswered questions or participant attrition. In the treatment of missing data, we will assess (and report) whether participants with missing data differ systematically from others on background or other characteristics, clinic or trial arm and consider this in the interpretation of findings. Depending on the findings of this assessment, we will adopt listwise deletion of missing cases with adjustments for covariates associated with missingness, multiple imputation or equivalent methods (eg, maximum likelihood estimates in mixed models).
Study limitations
Three potential limitations warrant discussion. First, we chose to implement the trial in 16 clinics in reasonable proximity to our research institution. This minimised costs and thus enabled us to enrol the recommended minimum number of clusters per trial arm.11 However, potential confounding from cluster effects may have been further minimised by a greater number of clinics and the racial, ethnic and linguistic diversity of participants enhanced by greater geographical diversity in clinics. Second, we chose to collect minimal contact information for participants eligible for the T2 and T3 surveys. Given the sensitivity of the topic, we considered this important for preventing both barriers to recruitment and risks to participants’ privacy. However, this simultaneously limited opportunities for participant contact and may compromise retention. Third, while clinic estimates of the total number of eligible patients during the study period will enable calculation of the participation rate, a lack of information on the characteristics of all eligible patients will preclude conclusions about sample representativeness.
Ethics and dissemination
Research ethics approval
Institutional Review Board approval for the study was granted by the Committee for the Protection of Human Subjects at Dartmouth College (Study #00028721) as well as by an external Institutional Review Board affiliated with one participating clinic.
Consent
When adopting a cluster randomised study design, it is not usually feasible to obtain participants’ consent to randomisation.11 47 48 Instead, consent to randomisation is typically provided by a surrogate decision-maker at the cluster level.47 48 We will obtain agreement to randomisation from a representative from each clinic and will seek eligible patients’ informed consent to participate in data collection for the study.
To eliminate barriers to participation and minimise risks to participants, we were granted Institutional Review Board approval for a waiver of documented informed consent and for a waiver of parental consent for participants aged 15–17 years. In our modified consent process, clinic staff will provide potentially eligible patients with a tablet computer (proactively and/or on request) that displays an electronic version of the study information sheet. This information sheet enables patients to self-assess their eligibility for the study, become informed about the study purposes, processes, benefits and risks and elect whether to participate in the study. Patients who select ‘Yes’ in response to the question ‘Now that you have read this information, do you agree to participate in this study?’ that follows this electronic information sheet will be taken as having given informed consent and will proceed to the T1 survey immediately. Paper copies of the information sheet will also be available in participating clinics. The study information sheet may be requested.
Privacy and confidentiality
We have adopted several strategies to protect the privacy of participants. We were granted Institutional Review Board approval for a waiver of documented informed consent (see Consent) and will administer the provision of participant compensation from the research institution so that no information on participants will be stored in any clinic. The waiver of documented informed consent also means that patients can participate in the study without ever providing their name or contact details if they are willing to forego compensation. Participants aged under 20 years may only elect to complete the T2 and T3 surveys online (with email communication) to safeguard their privacy. Participants aged 20 years and older who elect to complete the T2 and T3 surveys on paper will receive (and send) all associated correspondence in unbranded envelopes. We will use an otherwise meaningless random code generated during T1 survey completion to link participant surveys across the three time points. Finally, we have restricted access to identified participant-level data and files (see Data management).
Monitoring
Following internal institutional consultation, we determined that a formal Data and Safety Monitoring Board was unnecessary due to the minimal risks associated with study participation and the focus of the study on outcomes other than mortality or morbidity.49 We anticipate no harms arising from implementation of the interventions. If we become aware of any harms or other adverse events, either through study data or other avenues, we will review and address these according to standard institutional processes in consultation with the relevant Institutional Review Board(s). We will also file regular reports on trial progress and any adverse events with the Institutional Review Boards. Although we intend to conduct some analyses of pilot data collected before assigning clinics to trial arms, we do not anticipate undertaking any interim analyses of trial outcome data and have not devised any stopping guidelines.
Dissemination policy
Full protocol
Members of the scientific community and the public can access the full study protocol via this open access publication. Substantive modifications to this protocol will be communicated to the relevant staff at participating clinics during regular communications. Substantive modifications to this protocol will also be communicated to others via study meetings, written summaries, published modifications to the trial profile at clinicaltrials.gov and/or via statements in scientific papers or reports arising from the study.
Study findings
We will disseminate study findings through various channels. We will deliver presentations at scientific conferences and professional meetings and publish papers in peer-reviewed, open-access journals. We will adhere to International Committee of Medical Journal Editors recommendations pertaining to authorship roles and responsibilities in papers arising from this study.50 We will also prepare a final report of study findings and accompanying summary for lay audiences. We will disseminate these documents to participants and participating clinics, healthcare providers, policy makers, the public and other relevant groups. Wherever possible, we will make documents describing study findings available via the study website (http://www.rightforme.org). We do not intend to draw on the services of professional writers for the development of presentations, papers or reports.
Acknowledgments
We thank Amina Hetu and Nitzy Bustamante for their contribution to the development of study materials. We thank Shama Alam and Elizabeth Harman for participating in the production of study interventions. We also thank those who provided valuable feedback on early drafts of the Right For Me decision aids and other materials, including Amanda Beery, Rachel Darche, Ann Davis, Amanda Dennis, Candace Gibson, Christina Lachance, Lindsay Smith, Michele Stranger-Hunter, Lawrence Swiader, Christina Warner, Jacki Witt, Elisabeth Woodhams and Lauren Zapata.
References
Footnotes
↵i We intend to evaluate the feasibility and acceptability of the interventions from the perspectives of healthcare providers and other clinic staff in a separate, qualitative study.
↵ii The first iteration was: (1) What are my options? (2) What are the possible benefits and harms of those options? and (3) How likely are the benefits and harms of each option to occur?12 The second iteration was: (1) What are my options? (2) What are the possible benefits and harms of those options? and (3) How likely are each of those benefits and harms to happen to me?13
↵iii To enhance the usefulness of data for future research, participants who did not experience a contraceptive conversation will be asked to complete CollaboRATE with reference to the healthcare visit in general.
↵iv For participants who complete the T2 survey on the target completion date, the four-week recall period adopted for the assessment of contraceptive method(s) used in this survey will align with the four-week timeframe adopted in the assessment of intended contraceptive method(s) in the T1 survey.
Contributors RT, KZD and GE conceived the study. All authors contributed to the design of the study (eg, selection of interventions, development of participant and clinic eligibility criteria, selection of assignment approach, selection of study outcomes) and/or the development of study interventions, recruitment materials (eg, study posters, information sheets) and data collection materials and protocols (eg, patient surveys, survey invitation and reminder schedules). RT drafted the manuscript. All authors contributed to revisions of the draft and gave approval for publication of the manuscript.
Funding Research reported in this protocol was funded through a Patient-Centered Outcomes Research Institute (PCORI) Award (CDR-1403-12221). Apart from requiring adherence to Methodology Standards that specify best practices in the design and conduct of patient-centred outcomes research, the funder has had no role in study design, writing of the protocol or the decision to submit the report for publication.
Disclaimer The views presented in this protocol are solely the responsibility of the authors and do not necessarily represent the views of the Patient-Centered Outcomes Research Institute (PCORI), its Board of Governors or Methodology Committee. PCORI can be contacted at info@pcori.org.
Competing interests RT reports a grant from the Patient-Centered Outcomes Research Institute (PCORI) during the conduct of the study and non-financial support from PCORI outside the submitted work. RT also reports ownership of copyright in several patient decision aids and a role as an editor of the text, ‘Shared Decision Making in Health Care’ but has not received any personal income connected to this ownership or role. RM, KZD, GS, DA, TF, DJJ, ZL, ALO, and TDT report a grant from PCORI during the conduct of the study. MB and KKU report personal fees from Dartmouth College during the conduct of the study. MBB reports a grant from PCORI and other payments from Dartmouth College during the conduct of the study. PB reports personal fees and non-financial support from Dartmouth College during the conduct of the study and non-financial support from Dartmouth College outside the submitted work. CCB reports personal fees and non-financial support from Dartmouth College during the conduct of the study. JN reports personal fees from Dartmouth College during the conduct of the study and non-financial support from PCORI outside the submitted work. MN reports personal fees and other payments from Dartmouth College during the conduct of the study. MN also reports a role as a healthcare provider and clinic representative in a clinic participating in the study. HLS reports a role as a developer of the AskShareKnow programme intervention components and related survey items that were adapted for use in the study but has not received any personal income connected to this role. LFS reports personal fees from Dartmouth College during the conduct of the study; grants from Bayer Health Care Inc., Teva Pharmaceuticals and Gilead Pharmaceuticals Inc. outside the submitted work; and personal fees from Hologic Inc. outside the submitted work. LT reports other payments from Dartmouth College during the conduct of the study. GE reports a grant from PCORI during the conduct of the study and personal fees from Emmi Solutions LLC, Washington State Health Department, Oxford University Press, the National Quality Forum, SciMentum LLC, EBSCO Health, & think LLC and ACCESS Federally Qualified Health Centers outside the submitted work. GE also reports ownership of copyright in the CollaboRATE measure of shared decision-making, the Observer OPTION measure of shared decision-making and several patient decision aids.
Ethics approval The Dartmouth College Committee for the Protection of Human Subjects.
Provenance and peer review Not commissioned; externally peer reviewed.