Article Text

Abstract
Introduction Diabetes mellitus and diabetic nephropathy (DN) are prevalent and costly to manage. DN is the leading cause of end-stage kidney disease. Conventional therapy blocking the renin–angiotensin system has only achieved limited effect in preserving renal function. Recent observational data show that the use of Chinese medicine (CM), a major form of traditional medicine used extensively in Asia, could reduce the risk of end-stage kidney disease. However, existing clinical practice guidelines are weakly evidence-based and the effect of CM remains unclear. This trial explores the effect of an existing integrative Chinese–Western medicine protocol for the management of DN.
Objective To optimise parameters and assess the feasibility for a subsequent phase III randomised controlled trial through preliminary evaluation on the effect of an adjuvant semi-individualised CM treatment protocol on patients with type 2 diabetes with stages 2–3 chronic kidney disease and macroalbuminuria.
Methods and analysis This is an assessor-blind, add-on, randomised, controlled, parallel, multicentre, open-label pilot pragmatic clinical trial. 148 patients diagnosed with DN will be recruited and randomised 1:1 to a 48-week additional semi-individualised CM treatment programme or standard medical care. Primary end points are the changes in estimated glomerular filtration rate and spot urine albumin-to-creatinine ratio between baseline and treatment end point. Secondary end points include fasting blood glucose, glycated haemoglobin, brain natriuretic peptide, fasting insulin, C peptide, fibroblast growth factor 23, urinary monocyte chemotactic protein-1, cystatin C, nephrin, transforming growth factor-β1 and vascular endothelial growth factor. Adverse events are monitored through self-completed questionnaire and clinical visits. Outcomes will be analysed by regression models. Enrolment started in July 2015.
Ethics and registration This protocol is approved by the Institutional Review Board of the University of Hong Kong/Hospital Authority Hong Kong West Cluster (reference number UW 14-301).
Trial registration number NCT02488252.
- Chinese medicine
- glomerular filtration rate
- albuminuria
- clinical trial
- pragmatic
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Strengths and limitations of this study
This study is the first to assess the potential efficacy of an adjuvant Chinese medicine programme in diabetic kidney disease.
Chinese medicine is being extensively used in Asia and getting popular worldwide. Previous epidemiological data suggest that Chinese medicine may retard the deterioration of renal function among patients with diabetic kidney disease. Hence, it is timely to perform a clinical trial.
This study is a pilot study and the sample size is not powered to achieve statistical significance.
Since the trial is open-label, subjective outcomes including quality of life could not be assessed.
Introduction
The epidemic of diabetes mellitus (DM) is predicted to rise steadily in incidence and prevalence all over the world, driven by the effects of an ageing population and rising levels of obesity.1 ,2 It is estimated that 387 million people were having DM in 2014;3 and by 2030, the DM population is projected to reach 439 million.4 Diabetic nephropathy (DN) is the leading cause of end-stage kidney disease (ESKD)5 and is developed among one-third of all patients with diabetes.5 ,6 In Hong Kong, the incidence of ESKD primarily caused by DN requiring renal replacement therapy in the form of chronic dialysis or kidney transplantation doubled from 1996 (26.2%) to 2013 (49.6%).7 Patients with DN have the lowest adjusted 5-year survival probability of 50.6% when compared with patients with incident renal replacement therapy with other causes including hypertension (61.1%) and glomerulonephritis (71.1%).8 DN imposes a major burden on patients and healthcare systems in many developed regions.5
The pathogenesis of DN is diverse9–13 and yet to be fully understood. It involves glycotoxicity, advanced glycation end products and reactive oxygen species which induce haemodynamic changes and inflammation. Biopsy from patients with DN shows that podocyte loss, basement membrane thickening, interstitial fibrosis and tubular atrophy and DN are characterised by proteinuria clinically. Conventional therapy from the western literature pinpointing a few selected pathways, such as the widely advocated blockade of the renin–angiotensin system, has only achieved modest efficacy in treating DN.6 Combined therapy with several conventional regimes, for instance, ACE inhibitor and angiotensin receptor blocker may not be as efficacious as believed.6 ,14 No specific therapies for DN or renal fibrosis are available currently. Chinese medicine (CM) is a popular complementary remedy in addition to standard medical care among the Chinese in Hong Kong.15 ,16 A recent observational study in Taiwan shows that the use of CM is associated with an average risk reduction of 59% of ESKD when compared with non-users in a 6-year period. However, the risk reduction varies significantly across different subgroups of patients stratified clinically based on CM theory.17 Despite its popularity, there is no evidence-based clinical guideline for CM as an adjuvant therapy to standard medical care in managing DN.
An integrative Chinese–Western medicine clinical management protocol for DN was developed by reviews of existing clinical management guidelines and an expert consensus from CM nephrologists in mainland China in 2012.18 The adjuvant CM treatment consists of five different CM formulas corresponding to five subgroups of patients stratified by clinical manifestations. A preliminary evaluation on renal clinical outcomes among patients with DN attending an ongoing local CM service programme based on the captioned protocol shows stabilised glomerular filtration rate (GFR), reduced albuminuria and reduced glycated haemoglobin (HbA1c) level. Recent evidence from in vitro and in vivo studies suggested that the effect of these herbal medicines in reducing proteinuria and alleviating decline of renal function could be associated with attenuated podocyte apoptosis and detachment,19–21 reduced oxidative stress21 ,22 and suppressed transforming growth factor-β (TGF-β) production,23 ,24 through some of the important pathways including TGF-β/Smad,23 ,24 mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK)25 and nuclear factor kappa B (NF-κB)26 signalling pathway. In this pilot study, we aim to explore the effect of the captioned protocol on renal outcomes when used as an add-on therapy for patients with DN in the existing healthcare setting.
Objective
To optimise parameters and access the feasibility for a subsequent phase III randomised controlled trial (RCT) through preliminary evaluation on the effect of the adjuvant semi-individualised CM treatment protocol on patients with type 2 diabetes with stages 2–3 chronic kidney disease (CKD) and macroalbuminuria.
Method/design
Study design
This is an assessor-blind, add-on, randomised, controlled, parallel, multicentre, open-label pragmatic clinical trial. The flow of study is summarised in figure 1.
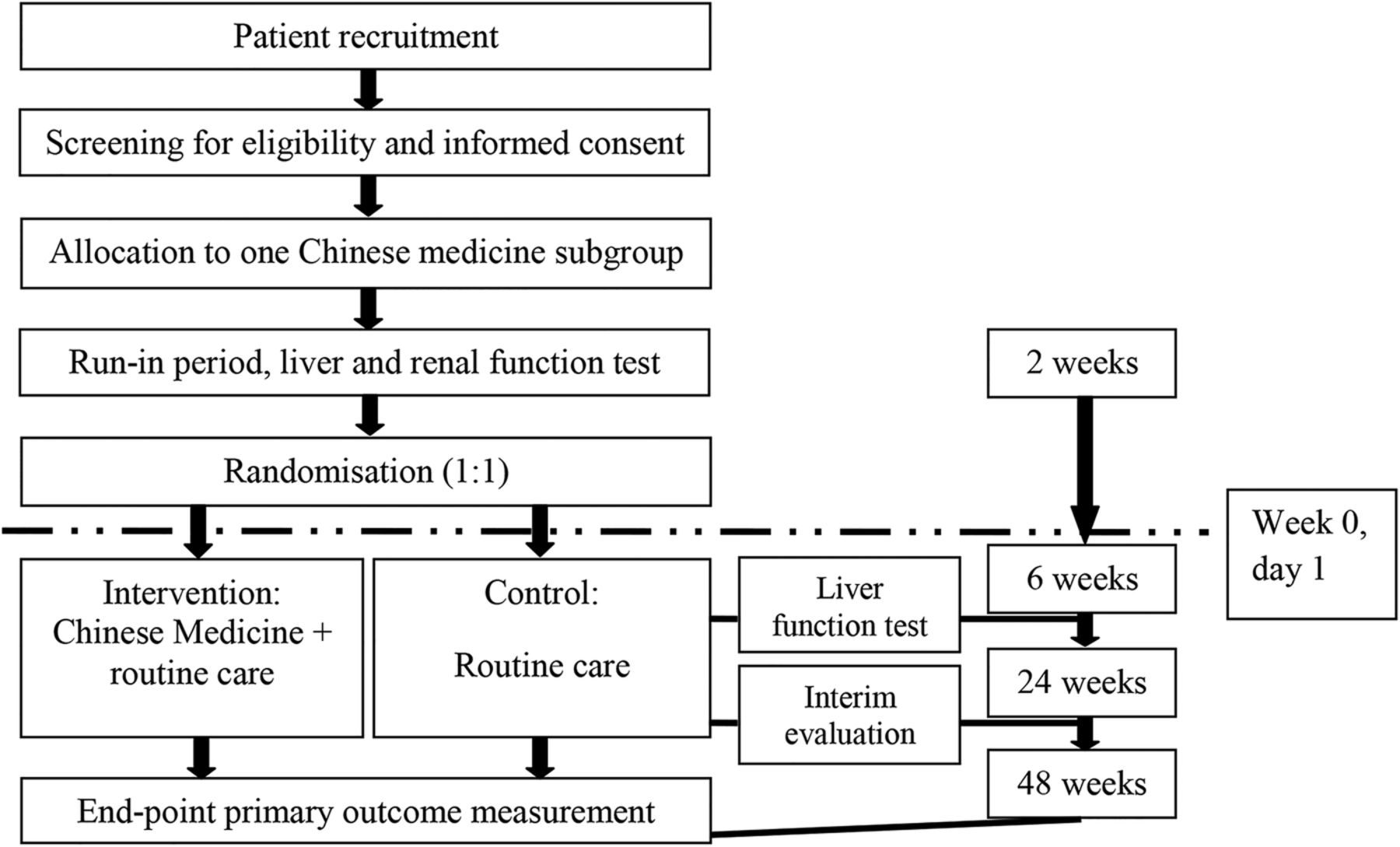
{kind=link}
Flow of study.
Inclusion and exclusion criteria
Patients: (1) diagnosed with type 2 diabetes for at least 5 years; (2) with an estimated GFR≥30<90 mL/min/1.73 m2 confirmed with repeat testing over 3 or more months calculated by the abbreviated Modification of Diet in Renal Disease (MDRD) study equation;27 (3) persistent macroalbuminuria with spot urine albumin-to-creatinine ratio (UACR) ≥300 mg/g confirmed by at least two consecutive first morning void urine samples; (4) aged between 35 and 80 years; (5) on a stable dose of antidiabetic drug including insulin for 12 weeks and (6) on a stable dose of ACE inhibitor or angiotensin receptor blocker for 12 weeks will be recruited.
Patients: (1) with a known history of glomerulonephritis, polycystic kidney disease, systemic lupus erythematosus or any suggestive evidence of non-diabetic glomerulopathy; (2) with a known history of kidney transplant; (3) with concurrent severe disorders of the heart, brain, liver and haematopoietic system, tumour, mental disorder; (4) deranged liver function; (5) with poorly controlled blood pressure; (6) with a known history of intolerance or malabsorption of oral medications; (7) uncontrollable urinary infection; (8) experiencing pregnancy and (9) participating in other clinical trials within 30 days will be excluded.
Sample size calculation
Currently, there is no consensus on the sample size calculation for pilot studies. Previous studies suggested a sample size of 12–30 per arm.28–31 Since the primary objective of this trial is to explore the feasibility of a subsequent main study and have a preliminary evaluation on the effect of CM on patients with DN, the sample size calculation is based on control of the inflation factor (IF) to the estimation of sample size for the subsequent main study.31
The standard deviation used for sample size calculation for the main study could be underestimated by a pilot study and should be adjusted by an IF.31 IF is calculated based on the size of the pilot study and the confidence level of achieving at least the desired power in the main study. The actual achieved power of the main study depends on the nominal power set for the main study and the IF.
In order to be 95% confident (two-sided) that the subsequent main study actually achieves a power of 80% with nominal power set at 90% (ie, a 10% power forfeit), the IF should be <1.15. At IF=1.15, a sample size of 80 is therefore needed to have 95% one-sided confidence that the main study will achieve at least the nominal power to test the hypothesis that an add-on of the whole CM treatment programme could be more effective in stabilising the GFR among patients with DN when compared with having standard care alone. For subgroup analysis, a sample size of 25 patients per subgroup could achieve 80% one-sided confidence that the effect of stabilising GFR is different within subgroups of similar CM clinical pattern.
With five subgroups, a sample size of 148 patients is needed in this pilot trial to allow a 15% attrition rate.
Recruitment and setting
Patients are recruited from Queen Mary Hospital Specialist Renal Outpatients Clinic, Sai Ying Pun Family Medicine Specialty Clinic, Sai Ying Pun Jockey Club General Out-patient Clinic, Kennedy Town Jockey Club General Out-patient Clinic, Aberdeen Family Medicine Specialty Clinic, Aberdeen Jockey Club General Out-patient Clinic; and Diabetes Complication Screening Clinic and Renal Clinic of Tung Wah Hospital. Patients diagnosed with diabetes and CKD stage 2–3 with macroalbuminuria are referred to the principal investigator (PI), co-investigator (Co-I), or Chinese medicine practitioner (CMP) for further assessment. The aim, procedures, nature of study, possible side effects of the CM treatment programme and right of unconditional withdrawal are explained by PI or Co-I before written consent is obtained from each patient who agrees to take part in the study.
All patients will undergo a 2-week run-in period, during which they are asked to keep their medication dosage fixed. A blood sample will be taken and tested to check liver function (γ-glutamyltransferase, alanine transaminase (ALT), alkaline phosphatase, total protein, aspartate transaminase (AST), bilirubin, albumin) and renal function (sodium, chloride, urea, potassium, creatinine) in the Department of Pathology & Clinical Biochemistry of Queen Mary Hospital or local clinical laboratories of other hospitals. Patients are considered eligible for the study if their liver functions are normal.
A random sequence is generated by and encrypted with computer by an independent research assistant (RA). Sealed opaque envelopes containing an allocation sequence are sent to the outpatient clinics. The password of the sequence is kept in a sealed, duly-signed opaque envelope locked by RA and Co-I. The allocation sequence is concealed from PI and Co-Is responsible for patient screening, recruitment and assignment of CM subgroup stratification. Eligible candidates are randomised to either the active treatment arm or control arm. The allocation will be explained by the CMP I or II to the patient. The allocation will be masked from the outcome assessor. The participant could not be masked due to the nature of treatment. Since the clinical outcomes under investigation are objective, placebo effect should be minimised. The distribution of known and unknown confounding factors among the intervention group and control group is balanced by the randomisation.
Intervention and control
The intervention under investigation is a semi-individualised CM treatment protocol used on top of standard medical care. The programme consists of five different CM herbal formulations corresponding to five different subgroups of patients stratified by clinical manifestations according to classical CM theory:
A, for the subgroup of ‘spleen and kidney Qi deficiency’:
Radix Aucklandiae (Mu Xiang), Rhizoma Pinelliae Praeparatum (Fa Ban Xia), Rhizoma Atractylodis Macrocephalae (Bai Zhu), Poria (Fu Ling), Radix et Rhizoma Glycyrrhizae Praeparata cum Melle (Zhi Gan Cao), Fructus Amomi (Sha Ren), Pericarpium citri reticulatae (Chen Pi), Radix et Rhizoma Ginseng (Ren Shen), Radix Rehmanniae Praeparata (Shu Di Huang), Fructus Corni (Shan Zhu Yu), Rhizoma Dioscoreae (Shan Yao), Cortex Moutan (Mu Dan Pi), Rhizoma Alismatis (Ze Xie).
B, for the subgroup of ‘spleen and kidney Yang deficiency’:
Rhizoma Atractylodis Macrocephalae (Bai Zhu), Fructus Chaenomelis (Mu Gua), Radix Aucklandiae (Mu Xiang), Fructus Tsaoko (Cao Guo), Semen Arecae (Bing Lang), Radix Aconti Lateralis Praeparata (Hei Shun Pian), Poria (Fu Ling), Rhizoma Zingiberis (Gan Jiang), Radix et Rhizoma Glycyrrhizae Praeparata cum Melle (Zhi Gan Cao), Rhizoma Zingiberis Recens (Sheng Jiang), Fructus Jujubae (Da Zao), Radix Rehmanniae Praeparata (Shu Di Huang), Fructus Corni (Shan Zhu Yu), Rhizoma Dioscoreae (Shan Yao), Cortex Moutan (Mu Dan Pi), Rhizoma Alismatis (Ze Xie), Ramulus Cinnamomi (Gui Zhi).
C, for the subgroup of ‘spleen and kidney Qi and Ying deficiency’:
Radix et Rhizoma Ginseng (Ren Shen), Radix Astragali (Huang Qi), Radix Rehmanniae Praeparata (Shu Di Huang), Fructus Corni (Shan Zhu Yu), Rhizoma Dioscoreae (Shan Yao), Poria (Fu Ling), Cortex Moutan (Mu Dan Pi), Rhizoma Zingiberis Recens (Sheng Jiang), Fructus Jujubae (Da Zao).
D, for the subgroup of ‘liver and kidney Ying deficiency’:
Radix Rehmanniae Praeparata (Shu Di Huang), Fructus Corni (Shan Zhu Yu), Rhizoma Dioscoreae (Shan Yao), Poria (Fu Ling), Cortex Moutan (Mu Dan Pi), Rhizoma Alismatis (Ze Xie), Fructus Ligustri Lucidi (Nv Zhen Zi), Herba Ecliptae (Mo Han Lian).
E, for the subgroup of ‘Ying and Yang deficiency’:
Radix Rehmanniae Praeparata (Shu Di Huang), Fructus Corni (Shan Zhu Yu), Rhizoma Dioscoreae (Shan Yao), Poria (Fu Ling), Cortex Moutan (Mu Dan Pi), Rhizoma Alismatis (Ze Xie), Ramulus Cinnamomi (Gui Zhi), Radix Aconti Lateralis Praeparata (Hei Shun Pian), Fructus Ligustri Lucidi (Nv Zhen Zi), Herba Ecliptae (Mo Han Lian).
Patients with diabetic kidney disease provided consent are further assessed by the CMP and categorised into one of the five different CM subgroups based on the Guiding Principle of Clinical Research on New Drugs of Chinese Medicine.32 The CM diagnoses are based on clinical manifestation as follows:
A, for the subgroup of ‘spleen and kidney Qi deficiency’:
Primary manifestation: fatigue, shortness of breath, torpid intake, lumbago.
B, for the subgroup of ‘spleen and kidney Yang deficiency’:
Primary manifestation: fear of cold, fatigue, shortness of breath, torpid intake, lumbago.
C, for the subgroup of ‘spleen and kidney Qi and Ying deficiency’:
Primary manifestation: fatigue, lumbago, dry mouth, heat vexation.
D, for the subgroup of ‘liver and kidney Ying deficiency’:
Primary manifestation: dizziness or headache, lumbago, dry mouth, heat vexation.
E, for the subgroup of ‘Ying and Yang deficiency’:
Primary manifestation: fear of cold, lumbago, dry mouth, heat vexation.
Patients with at least one episode of all the primary manifestations of each CM subgroup listed above during the 2 weeks before consultation are considered eligible for the corresponding group. To enhance the specificity of categorisation, patients having multiple manifestations that fit more than one subgroup are excluded.
The treatment programme is originated from an existing clinical practice guideline being used in mainland China derived from expert consensus.18 Patients randomised to the intervention arm will receive the corresponding formulations on top of standard medication, as stated above. Minor adjustment of the herbs is allowed based on an individual's condition and determined by the CMP to reflect actual clinical practice. All patients continue their standard medications and follow-ups. Patients randomised to the control group will receive consultation from the CMP at baseline, study midpoint (week 24) and study end point (week 48) in addition to standard care. Patients will have 5 days of medicine per week and are advised to take the medication twice per day dissolved in boiling water in the first 5 days of the week.
Herbal safety
Herbal medicines with known toxicity that are listed in Schedule 1, Chapter 549, Chinese Herbal Medicines, Chinese Medicine Ordinance of Hong Kong are excluded. Soluble herbal granules prepared by PuraPharm are used in this study. The production process is in strict compliance with standards of Good Manufacturing Practice (GMP). The CM is prescribed and dispensed through the clinics of the School of Chinese Medicine, Li Ka Shing Faculty of Medicine or Clinical Trial Pharmacy of Queen Mary Hospital complying with the standard of practice of pharmacy. A fully registered CMP is responsible for clinical diagnosis and prescription. At week 6, patients will undergo liver function tests and brain natriuretic peptide (BNP) test for monitoring of liver toxicity and cardiotoxicity. To further ensure patient safety, we only recruit patients with stages 2–3 CKD who are more clinically stable to participate in the study.
Outcome measurement
The primary outcome measures are the changes in the estimated GFR and spot UACR between the baseline (week 0) and treatment end point (week 48). Secondary measures include changes from baseline (week 0) to end of treatment (week 48) in fasting blood glucose (FBG), HbA1c, serum BNP, fasting serum insulin, serum C peptide, serum fibroblast growth factor 23 (FGF23), urinary monocyte chemotactic protein-1 (MCP-1), urinary cystatin C, urinary nephrin, urinary TGF-β1 and urinary vascular endothelial growth factor (VEGF). A self-complete questionnaire is distributed to the participants to monitor occurrence of adverse events, and they are advised to inform the PI or RA immediately.
Estimated GFR, UACR, FBG, HbA1c and liver function test are ordered by the Renal Clinic and assessed by the Division of Clinical Biochemistry, Queen Mary Hospital or local clinical laboratories of other hospitals which are accredited by the College of American Pathologists. BNP, insulin, C peptide, FGF23, MCP-1, cystatin C, nephrine, TGF-β1 and VEGF assessment will be performed at PI's research laboratory by an independent RA. Estimated GFR is calculated using the abbreviated 4-variable MDRD study equation with serum creatinine, age, ethnicity and gender. Clinical presentations and CM syndromic diagnosis will be assessed in a structured consultation developed for this purpose. To ensure consistency and reliability of assessment and minimise bias from investigators across the study, patients will be assessed by the same CMP during each visit based on standard CM practice. Follow-up consultations are held for all patients weekly in the first month and monthly subsequently. Evaluation of outcomes will be performed at weeks 24 (on treatment) and 48 (end of treatment). The follow-up schedule is summarised below in table 1.
Follow-up schedule
The baseline demographics including age, sex, body mass index, duration of diabetes, duration of diagnosed diabetic kidney disease and concurrent medications used are collected through the clinical management system of the Specialist Renal Outpatient Clinic. Patient flow measured as enrolled case per month and follow-up rate are recorded for the consideration of subsequent RCT design.
Handling of withdrawal and dropout
In order to maximise participants’ compliance, first, we have a thorough consent process for all participants with details of the study schedule, potential side effects of treatment, and the responsibilities of the participants explained. Second, we scrutinise potential participants carefully during a 2-week run-in period in order to exclude ineligible and low compliance participants before randomisation. Third, an independent email account and a direct telephone line associated with this clinical trial have been set up to enable active communication with patients. Moreover, extra visits will be arranged for patients if necessary. If any patient plans to withdraw or drop out, the study team will communicate with the patient to determine and resolve the problem in order to retain the participant.
Termination criteria
The trial will be terminated for a specific participant if she or he has: (1) presence of serious adverse effect(s) in the interventional group; (2) hypersensitivity towards Chinese herbal medicine and (3) participation in another clinical trial. The whole study will be terminated under the following circumstances: (1) presence of serious adverse effect(s) related to Chinese herbal medicine with supportive evidence and (2) completion of all follow-up assessments.
Serious adverse events include adverse events that result in death, require either hospitalisation or the prolongation of hospitalisation, are life-threatening, result in a persistent or significant disability/incapacity or result in a congenital anomaly/birth defect. Other important medical events, based on appropriate medical judgement, may also be considered serious adverse events if a patient's health is at risk and intervention is required to prevent an outcome mentioned.
Data analysis
Missing values, if any, will be imputed with regression. The analysis will follow the intention-to-treat approach. Patients without a postrandomisation assessment for a particular efficacy end point will be excluded from the analysis of that end point. SAS and SPSS will be used for the data analysis.
The demographics of each group will be presented as means. Sex will be presented as the male:female ratio. Regression analyses will be used to compare the adjusted mean of estimated GFR, UACR, HbA1c, FBG, BNP, insulin, C peptide, FGF23, MCP-1, cystatin C, nephrin, TGF-β1 and VEGF at week 48 between (1) the combination of all intervention groups and combination of all control groups, (2) individual treatment subgroup and its matching control group and (3) different control groups with the corresponding baseline values as covariates. Data will be presented as the difference in adjusted means between the intervention arm and control arm with a 95% CI and the corresponding p value. A change score analysis will be performed as supplementary analysis.
The adverse events are recorded based on the Common Terminology Criteria for Adverse Events V.4.03 and will be analysed in a narrative manner. The adverse events are categorised into five grades: grade (1) mild; asymptomatic or mild symptoms; clinical or diagnostic observations only; no intervention indicated; grade (2) moderate; minimal, local or non-invasive intervention indicated; limiting age-appropriate instrumental activities of daily living; grade (3) severe or medically significant but not immediately life-threatening; hospitalisation or prolongation of hospitalisation indicated; disabling; limiting self-care activities of daily living; grade (4) life-threatening consequences; urgent intervention indicated; and grade (5) death related to adverse events. The percentage of all adverse events and the rate of attrition due to adverse events will be compared between intervention groups and control groups.
To minimise type I error inflation, the analysis will follow a hierarchical approach in the order of (1) individual treatment subgroup versus its control subgroup and (2) combination of all intervention groups versus the combination of all control groups as to avoid type I error inflation. The primary outcomes are the change of GFR and UACR.
Subgroup analysis will be performed for CKD stages 2 and 3 separately. Sensitivity analyses will be performed for (1) missing data imputed with regression, (2) missing data imputed with last-observation-carried-forward (LOCF) and (3) per protocol dropout of patients. Adverse events will be analysed case-by-case and presented descriptively.
Data management
A trial management committee is formed by the study team members. Co-Is and RA collect, clean and send the study data to the committee on a weekly basis. All data are double-entered to computer and cleaned before analysis to prevent data entry errors. All data transfer is encrypted to protect patients’ confidentiality. The committee holds meetings monthly with experts to discuss the progress of the trial.
Ethics consideration
This trial is ethically justified as there are uncertainties on the therapeutic effect and adverse effect of the treatment programme which is generated by expert consensus but with no further validation of clinical evidence. Since the treatment is being used in mainland China and has the potential to be broadly used in Hong Kong and benefit local citizens, we believe this trial is worthwhile. All protocol amendments will be approved by the Institutional Review Board prior to execution.
References
Footnotes
Contributors KWC and SCWT conceived and designed the study. TPI, ASKK, SLL, GCWC, KCBT and YF contributed to the trial design and study operation. BJC contributed to the statistical design and analysis. WHY, DWLW, YL, JCKL, LYYC and KNL contributed to the design and operation of biochemical sampling. All the authors made significant contributions to the development and conceptualisation of the protocol. All the authors reviewed the draft versions of this paper and have read and approved the final manuscript.
Funding This study is conducted as an investigator-initiated study with financial support from the Health and Medical Research Fund (project number 12133341) administered by the Food and Health Bureau, the Government of the Hong Kong Special Administrative Region.
Disclaimer The funding agency has no role in the design of this study and will not have any role during its execution, analyses, interpretation of the data or decision to submit results.
Competing interests None declared.
Patient consent Obtained.
Ethics approval This protocol is approved by the Institutional Review Board of the University of Hong Kong/Hospital Authority Hong Kong West Cluster (HKU/HA HKW IRB; reference number UW 14-301). A WHO Trial Registration Data Set is presented below.⇓
Provenance and peer review Not commissioned; externally peer reviewed.