Article Text

Abstract
Introduction Pre-eclampsia (PE) affects 2–3% of all pregnancies and is a major cause of maternal and perinatal morbidity and mortality. Prophylactic use of low-dose aspirin in women at risk for PE may substantially reduce the prevalence of the disease. Effective screening for PE requiring delivery before 37 weeks (preterm PE) can be provided by a combination of maternal factors, uterine artery Doppler, mean arterial pressure, maternal serum pregnancy-associated plasma protein A and placental growth factor at 11–13 weeks' gestation, with a detection rate of 75% at a false-positive rate of 10%. We present a protocol (V.6, date 25 January 2016) for the ASpirin for evidence-based PREeclampsia prevention (ASPRE) trial, which is a double-blinded, placebo-controlled, randomised controlled trial (RCT) that uses an effective PE screening programme to determine whether low-dose aspirin given to women from 11 to 13 weeks' gestation will reduce the incidence of preterm PE.
Methods and analysis All eligible women attending for their first trimester scan will be invited to participate in the screening study for preterm PE. Those found to be at high risk of developing preterm PE will be invited to participate in the RCT. Further scans will be conducted for assessment of fetal growth and biomarkers. Pregnancy and neonatal outcomes will be collected and analysed. The first enrolment for the pilot study was in April 2014. As of April 2016, 26 670 women have been screened and 1760 recruited to the RCT. The study is registered on the International Standard Randomised Controlled Trial Number (ISRCTN) registry.
Trial registration number ISRCTN13633058.
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Strengths and limitations of this study
This is the largest multicentre, double-blinded, randomised placebo-controlled trial to examine the effect of aspirin in women who are high risk of developing pre-eclampsia.
The screening will occur in the first trimester as to allow for the maximum benefit of aspirin.
One hundred and fifty milligrams of aspirin will be used to reduce the incidence of aspirin resistance and maximise the effect.
Follow-up of the offspring is limited to the early postnatal phase.
Background
Pre-eclampsia (PE) is an important cause of maternal and perinatal mortality and morbidity. There is extensive evidence that the risk of adverse outcome in relation to PE is much higher when the disease is severe and of early onset requiring delivery before 37 weeks' gestation (preterm PE), than at term.1–4 A major challenge in modern obstetrics is early identification of pregnancies at high risk of preterm PE and undertaking the necessary measures to improve placentation and reduce the prevalence of the disease.
Prediction of preterm PE
Extensive research in the past 20 years, mainly as a consequence of the shift in screening for aneuploidies from the second to the first trimester of pregnancy, has identified a series of early biophysical and biochemical markers of impaired placentation.5 A combination of maternal demographic characteristics, including medical and obstetric history, uterine artery pulsatility index (PI), mean arterial pressure (MAP) and maternal serum pregnancy-associated plasma protein-A (PAPP-A) and placental growth factor (PlGF) at 11–13 weeks' gestation can identify a high proportion of pregnancies at high risk for PE.6 A recent study involving 58 800 singleton pregnancies examined at 11–13 weeks’ gestation has further refined the prediction algorithm for PE. Using this algorithm, the estimated detection rate of preterm PE was 75%, at a false-positive rate of 10%.6
Prevention of preterm PE
The prophylactic use of low-dose aspirin for prevention of PE has been an important research question in obstetrics for the past three decades. In 1979, Crandon and Isherwood7 observed that nulliparous women who had taken aspirin regularly during pregnancy were less likely to have PE than those who did not. There have been two meta-analyses published reporting that the administration of low-dose aspirin in high-risk pregnancies is associated with a decrease in the rate of PE.8 ,9 However, there are also other possible pathways which lead to the development of PE among different risk groups, and it is not known which risk factors or pathological processes may be responsive to early initiation of low-dose aspirin.
Initiation of low-dose aspirin in early pregnancy
In most studies that evaluated aspirin for the prevention of PE, the initiation of treatment was at or after 16 weeks' gestation. Examination of a small number of randomised trials of low-dose aspirin in women at high risk for PE suggests that the effectiveness of therapy is related to the gestational age at the initiation of treatment. A meta-analysis by Bujold et al9 reported that low-dose aspirin started at 16 weeks or earlier was associated with a significant reduction in the relative risk (RR) for PE (0.47, 95% CI 0.34 to 0.65) and fetal growth restriction (FGR; 0.44, 95% CI 0.30 to 0.65). In contrast, aspirin started after 16 weeks did not have a significant benefit (PE: RR 0.81, 95% CI 0.63 to 1.03; FGR: RR 0.98, 95% CI 0.87 to 1.10). More detailed analyses of these data on PE demonstrated that low-dose aspirin started at or before 16 weeks' gestation was particularly effective in preventing preterm PE rather than term PE (RR: 0.11, 95% CI 0.04 to 0.33 vs RR: 0.98, 95% CI 0.42 to 2.33).10
The small number and small size of individual trials preclude definitive conclusions to be drawn regarding the effectiveness of aspirin starting before 16 weeks and the results need to be examined in a prospective major randomised trial.
Aspirin resistance
There is evidence that ∼30%, 10% and 5% of pregnant women are ‘aspirin resistant’ with dosage of 81, 121 and 162 mg, respectively.11 Furthermore, a retrospective cohort study reported that women who were identified by the PFA-100 test as being resistant to 81 mg of aspirin were less likely to develop severe PE when the dose of aspirin was increased from 81 to 162 mg, compared with those who continued with 81 mg.12 Consequently, a trial investigating the effectiveness of low-dose aspirin in the prevention of preterm PE should use a dose closer to 160 than 80 mg.
Safety of low-dose aspirin
The relative safety of first trimester use of low-dose aspirin has been demonstrated in large cohort and case–control studies, which reported that the drug is not associated with increase in risk of congenital heart defects or other structural or developmental anomalies.13–16
Randomised studies reported that ∼10% of women receiving low-dose aspirin presented with gastrointestinal symptoms; however, there was no evidence of increase in any type of maternal bleeding.17–19 Similarly, the best evidence suggests that low-dose aspirin started before 16 weeks' gestation does not increase the risk of placental abruption (RR 0.62, 95% CI 0.08 to 5.03).9 No additional adverse effects related to epidural anaesthesia have been reported in women taking low-dose aspirin compared with those taking placebo.20
Prospective and case–control studies did not find an association between daily consumption of 60–150 mg of aspirin during the third trimester and antenatal closure of the ductus arteriosus.21–23 A meta-analysis including more than 26 000 women randomised to low-dose (80–150 mg) aspirin or placebo/no treatment during pregnancy demonstrated that the use of aspirin was not associated with an increase in intraventricular haemorrhage or other neonatal bleeding.24 On the basis of currently available evidence, it would be reasonable to continue with low-dose aspirin well into the third trimester of pregnancy.
Hypothesis
We hypothesise that prophylactic low-dose aspirin administered from first trimester of pregnancy in women at increased risk for preterm PE will reduce the incidence and severity of the disease.
Aim
To examine if the prophylactic use of low-dose aspirin administered from the first trimester of pregnancy in women at increased risk for preterm PE can reduce the incidence and severity of the disease.
Objectives
Primary objective
To determine the efficacy of low-dose aspirin (150 mg daily), given to high-risk women from 11–14 to 36 weeks' gestation, in reducing the incidence of preterm PE, requiring delivery before 37 weeks.
Secondary objectives
To determine the effect of low-dose aspirin on adverse outcome of pregnancy at <37 weeks.
PE requiring delivery at <37 weeks;
Small for gestational age (<5th centile) requiring delivery at <37 weeks;
Miscarriage or stillbirth at <37 weeks;
Placental abruption (clinically or on placental examination) at <37 weeks;
Composite of any of the above.
To determine the effect of low-dose aspirin on adverse outcome of pregnancy at <34 weeks.
PE requiring delivery at <34 weeks;
Small for gestational age (<5th centile) requiring delivery at <34 weeks;
Miscarriage or stillbirth at <34 weeks;
Placental abruption (clinically or on placental examination) at <34 weeks;
Composite of any of the above.
To determine the effect of low-dose aspirin on adverse outcome of pregnancy at >37 weeks.
PE requiring delivery at >37 weeks;
Small for gestational age (<5th centile) requiring delivery at >37 weeks;
Miscarriage or stillbirth at >37 weeks;
Placental abruption (clinically or on placental examination) at >37 weeks;
Composite of any of the above.
To determine the effect of low-dose aspirin on neonatal mortality and morbidity.
Neonatal intensive care unit admission;
Intraventricular haemorrhage (IVH) grade II or above—defined as bleeding into the ventricles
Grade II (moderate)—IVH occupies <50% of the lateral ventricle volume,
Grade III (severe)—IVH occupies >50% of the lateral ventricle volume,
Grade IV (severe)—haemorrhagic infarction in periventricular white matter ipsilateral to a large IVH;
Ventilation—defined as need of positive pressure (continuous positive airway pressure (CPAP) or nasal CPAP) or intubation;
Neonatal sepsis—confirmed bacteraemia in cultures;
Anaemia—defined as low haemoglobin and/or haematocrit requiring blood transfusion;
Respiratory distress syndrome—defined as need of surfactant and ventilation as a result of prematurity;
Necrotising enterocolitis (NEC) requiring surgical intervention;
NEC is defined by a combination of clinical, radiological and laboratory features:
Systemic signs—apnoea, bradycardia, temperature instability, hypotension.
Intestinal signs—abdominal distension, gastric residuals, bloody stools, absent bowel sounds, abdominal tenderness, peritonitis.
Radiological signs—pneumatosis intestinalis or portal venous air, pneumoperitoneum.
Laboratory changes—metabolic and or respiratory acidosis, thrombocytopaenia, disseminated intravascular coagulation.
Composite of any of the above.
To determine the effect of low-dose aspirin on the incidence of neonatal birth weight below the 3rd, 5th and 10th centiles.
Birth weight will be recorded in the participants' medical notes, and birthweight percentile for gestational age at delivery is calculated using a normal range derived from our population.25
To determine the effect of low-dose aspirin on the incidence of stillbirth or neonatal death.
Owing to any cause;
Ascribed to PE or FGR;
In association with maternal or neonatal bleeding.
To determine the effect of low-dose aspirin on the incidence of spontaneous preterm delivery at <34 and <37 weeks.
Spontaneous delivery at <34 weeks (early preterm) and at <37 weeks (total preterm) includes those with spontaneous onset of labour and those with preterm prelabour rupture of membranes.
Centres
There are 13 academic hospitals participating in the trial. There are six centres in the UK; three in Spain; and one in each of Milan, Brussels, Greece and Israel.
Design
There are three components to the study: an internal pilot study, a screening quality study and a screening study followed by a double-blinded randomised placebo-controlled trial. Informed consent will be obtained by a trained healthcare professional who is a member of the study team at each particular centre.
Internal pilot study
The main study has been preceded by a 2-month pilot study, undertaken at King's College Hospital. In total, 1106 participants have been consented into the screening study and 56 participants to the randomised controlled trial (RCT). This pilot study has been used to assess the feasibility of recruitment to the screening study and RCT, and the ability of the centre to ensure successful compliance. A review by the ASpirin for evidence-based PREeclampsia prevention (ASPRE) Independent Data Monitoring Committee (IDMC) and Trial Steering Committee (TSC) of the internal pilot study has demonstrated the study has been successful with respect to recruitment to the screening study and RCT; however, it has also highlighted the complexity of the main ASPRE trial and confirmed the need for enhanced quality systems to be in place in advance of starting the main ASPRE trial in order to ensure the quality of pivotal data.
Screening quality study
A screening quality study, with a minimum recruitment period of 1–3 months (dependent on sites' performance) at each site, has been introduced to precede the main ASPRE trial. The aim of this study is to establish systems that will monitor quality of the measurement of uterine artery PI, MAP, PAPP-A and PlGF in a more detailed, formalised manner at sites and use these systems to assess quality, identify areas for improvement and, where required, implement strategies to improve quality, for example, retraining. This is based on the Down's syndrome screening quality assurance support service (DQASS) system that has been successful for improving the quality of the ultrasound and biochemical measurements in the National Health Service (NHS) fetal anomaly screening programme.
Recruitment rates have also been monitored. Furthermore, an assessment of data quality was made by the trial team at University College London Comprehensive Clinical Trials Unit (UCL CCTU) and any site-specific operational issues, which could not have been foreseen by the site assessment process, were identified, and addressed in advance of starting the main ASPRE trial.
Screening study and RCT
Following receipt of the results of the screening study, eligible high-risk women will be invited to take part in the RCT designated by the trial teams. It is anticipated that 10% of the population will screen positive for preterm PE and be invited to participate in the trial (figure 1).

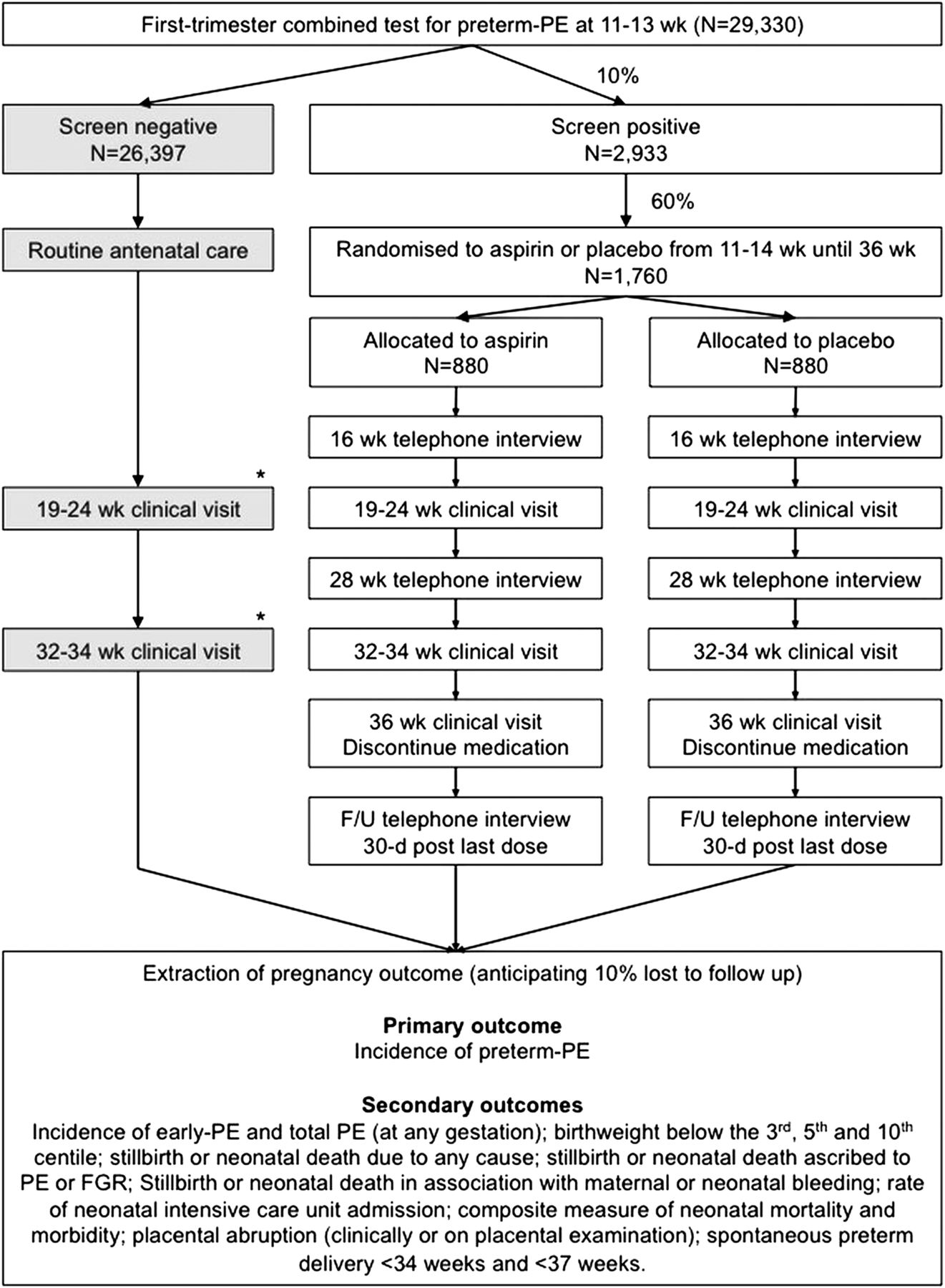{kind=link}
Flow chart of participants in the screening study and the randomised controlled trial. *Clinical visits at 19–24 and 30–37 weeks will only be performed on screen-negative participants at sites where a scan is performed by the fetal medicine unit as part of the routine clinical care pathway at either of these times. FGR, fetal growth restriction; PE, pre-eclampsia.
Inclusion and exclusion criteria
Screening phase inclusion criteria
Age >18 years;
Singleton pregnancy;
Live fetus at 11–13 weeks of gestation;
English, Italian, Spanish, French, Dutch or Greek speaking (otherwise interpreters will be used);
Informed and written consent.
Screening phase exclusion criteria
Multiple pregnancy;
Pregnancies complicated by major fetal abnormality identified at the 11–13 weeks assessment;
Women who are unconscious or severely ill, those with learning difficulties, or serious mental illness;
Age <18 years.
Randomisation inclusion criteria following screening
Screening phase inclusion criteria fulfilment;
High risk for preterm PE at 11–13 weeks by the algorithm combining maternal history and characteristics, biophysical findings (MAP and uterine artery PI) and biochemical factors (PAPP-A and PlGF);
Randomisation exclusion criteria following screening
Women taking low-dose aspirin regularly;
Bleeding disorders such as Von Willebrand's disease;
Peptic ulceration;
Hypersensitivity to aspirin or already on long-term non-steroidal anti-inflammatory medication;
Concurrent participation in another drug trial or at any time within the previous 28 days;
Any other reason the clinical investigators think will prevent the potential participant from complying with the trial protocol.
Methods
We will recruit women attending for their routine first scan in pregnancy at 11–13 weeks' gestation in the UK, Spain, Belgium, Italy, Greece and Israel. All eligible women attending for their routine first scan in pregnancy at 11–13 weeks' gestation are invited to take part. For the screening quality study and the screening study, the patient information sheet (PIS) will be sent with the appointment letter to all potential participants.
In women who agree to participate in the screening quality study, after obtaining informed consent, we measure the maternal MAP by automated devices,26 use transabdominal colour Doppler ultrasound to visualise the left and right uterine artery and measure the PI in each vessel and calculate the mean PI.27 Maternal serum PlGF is measured in the same blood sample taken for the measurement of PAPP-A, using automated machines that provide reproducible results (DELFIA Xpress system, PerkinElmer Life and Analytical Sciences, Waltham, Massachusetts, USA) as part of the routine screening for Down's syndrome. Participants enrolled in the screening quality study will not be informed of their risk of developing PE and will be managed according to routine standard of care at the site they attend. The principal investigators at each site are doctors who received their training by KHN and follow the Fetal Medicine Foundation (FMF) guidelines on how to undertake the appropriate measurements.
In women who agree to participate in the screening study of the main trial, after obtaining informed consent, we measure maternal MAP, uterine artery PI, PAPP-A and PlGF as described above. Following screening for preterm PE, high-risk women will be invited to take part in the RCT by designated members of the trial teams. Women eligible to participate in this trial will receive written information on the test drug and provide informed consent. When randomised, participants will be assigned a randomisation code. The randomisation codes will determine who receives placebo or aspirin 150 mg. The investigational medicinal product (IMP) supplier (Mawdsley Brooks and Co) will keep and store the randomisation code list. All participants, the principal investigator and clinical trial pharmacy will remain blind to trial drug allocation.
Data collection
Participant data for this study will be entered into an electronic case report form (CRF). For participants in the RCT the CRF will be printed and signed by the enrolling researcher.
Randomisation
Randomisation will be performed using a web-based system Sealed Envelope. The website randomly assigns participants to a randomisation code which corresponds to treatment packs with the same code at a given site. Each treatment pack will only be identified by a randomisation code. The treatment allocation will only be revealed to the researchers after completion of the study or where clinically essential.
Concealment of allocation
Mawdsley Brooks and Co will provide labelling (for all cartons and blister sheets) ensuring complete blinding of the IMP to all investigators and participants in the study, which includes the principal investigator, participating research doctors, pharmacists at the local clinical trial pharmacy, project managers and others involved in the trial. They are all blinded to the IMP allocation. Matching placebo tablets will be identical to the intervention (aspirin) in such parameters as size, thickness, physical properties and appearance. A film coating will be applied to the placebo tablets for aesthetic and taste reasons.
Mawdsley Brooks and Co will keep the randomisation code list confidential to maintain the blind; however, the randomisation code list will be transferred to Sealed Envelope to enable online randomisation and unblinding service to be established.
Intervention
Participants will take one tablet per night of either aspirin 150 mg or matched placebo. Participants will be asked to stop taking tablets at 36 weeks' gestation or, in the event of early delivery, at the onset of labour (maximum duration of 25 weeks). The aspirin tablets will be film-coated, to be taken orally once per night from enrolment until 36 weeks' gestation.
Study assessment
The study procedure by visit has been outlined in table 1.
Summary of the study visits
Laboratory tests
At the time of the 11–13 weeks scan, 20 mL of maternal blood will be taken for the measurement of PAPP-A and PlGF using automated machines that provide reproducible results (DELFIA Xpress system, PerkinElmer Life and Analytical Sciences, Waltham, Massachusetts, USA). The remaining serum and plasma will be stored at −80°C for future studies of potential biochemical markers for adverse pregnancy outcomes.
Participant compliance
Participants will be asked to bring their trial medication to each clinical visit; IMP compliance will be assessed by trial teams by counting remaining tablets at each follow-up visit and asking about compliance at telephone follow-up. Compliance with other aspects of the trial protocol will also be assessed. Participants will be encouraged to report any concerns or side effects in a diary for review at each trial visit.
Outcomes
Primary outcome
Incidence of preterm PE (delivery at <37 weeks).
PE will be defined as per the International Society for the Study of Hypertension in Pregnancy.29 The systolic blood pressure should be 140 mm Hg or more and/or the diastolic blood pressure should be 90 mm Hg or more on at least two occasions 4 hours apart developing after 20 weeks of gestation in previously normotensive women (blood pressure <140/90 mm Hg) and there should be proteinuria of 300 mg or more in 24 hours or urinary protein creatinine ratio of 30 mg/mmol or more or two readings of at least ++ on dipstick analysis of midstream or catheter urine specimens if no 24-hour collection is available. The efficacy will be assessed by the development of PE at any gestation after 20 weeks of pregnancy as defined above.
Secondary outcomes
As defined above in the Secondary objectives section.
Collection of pregnancy and neonatal outcomes
Data on pregnancy and neonatal outcomes will be collected from the hospital maternity records or their general medical practitioners. The obstetric records of the randomised women with pre-existing or pregnancy-associated hypertension will be examined to determine if the condition was chronic hypertension, PE or gestational hypertension. In the event, neonates are admitted to special care baby unit (SCBU), additional neonatal outcomes will be collected from the discharge summary of SCBU.
Side effects and adverse events reporting
Adverse event (AE) and reaction (AR) data are not being collected for participants of the screening quality study, or the screen-negative participants in main ASPRE RCT, as they are non-Clinical Trial of an Investigational Medicinal Product (CTIMP), which do not expose participants to any additional risk over and above that of routine clinical care.
Safety evaluations will be conducted at each of the RCT participants' follow-up visits. AEs include any unwanted side effects, sensitivity reactions, abnormal laboratory results, injury or intercurrent illnesses, and may be expected or unexpected. The period for AE reporting will be from the time of first dose until 30 days postfinal IMP administration. The participants will be followed up by a telephone interview 30 days after the last dose of IMP. These AEs will be recorded on the electronic CRF and do not need to be reported to the sponsor. The participants are instructed to contact a member of the trial team if there are any concerns regarding their medication.
Serious AEs/ARs (SAEs/SARs) occurring in the mother or baby from the time a participant is randomised until 30 days after stopping taking the IMP or until 30 days after delivery or until 30 days after the estimated due date, respectively, whichever is later, will be reported to the sponsor using the trial documentation. The standard definition of an SAE will be used.30
For the purposes of this study, the following events are included as protocol-defined exceptions to SAE reporting should only be reported to the sponsor as an SAE/SAR if the investigator believes the event is a result of the ASPRE intervention: hospitalisation for maternal or fetal observation, including minor bleeding episodes; preterm delivery (spontaneous, for maternal or fetal indication); miscarriage; stillbirth or neonatal death; admission of baby to neonatal intensive care unit; termination for fetal or maternal indication. If the event is deemed to be part of the routine progress of the pregnancy concerned, these events should be reported to the sponsor as a protocol-defined exception to SAE reporting, within the respective reporting timelines.
Statistical analysis plan including sample size and power calculation
The sample size calculation is based on a 76% detection rate of the first trimester combined screening for preterm PE at a screen-positive rate of 10%. With the aim to achieve a significant 50% reduction in the prevalence of preterm PE from 7.6% in the placebo group to 3.8% in the aspirin group, with a power of 90%, and 5% significance level, it is necessary to randomise 1600 high-risk pregnancies. If we allow for 10% loss to follow-up, it will be necessary to randomise a total of 1760 high-risk pregnancies, 880 women in each of the aspirin and placebo arms. On the assumption that 60% of high-risk pregnancies will agree to randomisation, we need to identify 2933 high-risk pregnancies (which will constitute 10% of the screened population). We will therefore have to recruit a total of 29 330 pregnancies to the screening study.
Type of analysis and statistical tests
The primary analysis will be a mixed-effects logistic regression analysis of the incidence of preterm PE with fixed effects for treatment and risk, and random effects for centre. A choice of transformation of risk (eg, logit transform) or grouping into levels will be made on the basis of a blinded review of the data. The treatment effect will be tested at the two-sided 5% level. Ninety-five per cent CIs will be produced for the proportions developing preterm PE in each of the two groups and for the treatment effect (active–placebo).
Planned secondary analysis of the primary outcome will include a survival analysis of the time to delivery with PE treating births for other causes as censoring. Prespecified baseline variables considered to be predictive will be included as appropriate. Their interactions with the treatment effect will be investigated. Gestational age at randomisation and its interaction with treatment will also be investigated. This analysis of treatment interactions will be considered as exploratory.
Descriptive statistics
A full set of descriptive statistics for all variables, overall and by treatment group, will be produced. Graphical displays will be produced as appropriate.
Secondary analysis
Secondary outcomes will be compared across treatment groups using appropriate tests. p Values and 99% CIs will be produced for treatment effects. No corrections will be made for multiplicity.
Safety
The incidence rates of AEs and SAEs and their relationship to trial drugs will be summarised by treatment group. The proportion of women discontinuing treatment will be summarised by reason and by treatment group.
Committee oversights
The IDMC is independent from the trial and is responsible for monitoring the progress of the trial including recruitment, protocol adherence, SAEs and side effects of treatment as well as the difference between the trial treatments on the primary outcome measures. They are the only oversight body that has access to unblinded accumulating comparative data. The IDMC is responsible for safeguarding the interests of trial participants, monitoring the accumulating data and making recommendations to the TSC on whether the trial should continue as planned.
The TSC is the independent group responsible for oversight of the trial in order to safeguard the interests of trial participants. The TSC provides advice to the chief investigator, co-chief investigator, UCL CCTU, the funder and sponsor on all aspects of the trial through its independent chair.
Ethics and dissemination
The study will be conducted in accordance with the principles of Good Clinical Practice. A favourable ethical opinion was obtained from London-Fulham Research Ethics Committee, reference number 13/LO/1479. Subsequent approval by individual ethical committee and competent authority was granted. Results will be published in peer-reviewed journals and disseminated at international conferences.
Discussion
The traditional approach to screening for PE is to identify risk factors from maternal demographic characteristics and medical history, but such an approach can identify only 35% of total PE and about 40% of preterm PE at false-positive rate of about 10%.31 ,32
In a proposed new approach to antenatal care, the potential value of an integrated clinic at 11–13 weeks' gestation in which maternal characteristics and history are combined with the results of a series of biophysical and biochemical markers to assess the risk for a wide range of pregnancy complications has been extensively documented.33 Effective screening for preterm PE can be achieved in this clinic with a detection rate of about 76% at a false-positive rate of 10%.6 There is a suggestion that the prevalence of PE can be halved by prescribing pregnant women low-dose aspirin before 16 weeks' gestation,9 and by using an enhanced screening approach using maternal demographics and history with biochemical and biophysical markers, these women can be identified effectively and entered into a double-blinded randomised placebo-controlled trial to assess whether low-dose aspirin can truly reduce the prevalence of preterm PE when given in the first trimester of pregnancy.
References
Footnotes
Contributors LCP and KHN conceived and designed the study. LCP, KHN, NO and DLR drafted the original grant proposal and trial protocol. DW provided methodological and statistical expertise. LCP and KHN provide expertise in the pregnancy clinical outcomes. LCP, KHN, NOG, DLR and the clinical project manager drafted the original protocol. LCP and NO drafted the manuscript. LCP, NO, DLR, with the support of the trial manager and the clinical project manager, have responsibilities for day-to-day running of the trial including participant recruitment, data collection and liaising with other sites. All authors critically reviewed and approved the final version of the manuscript.
Funding This study is supported by grants from the European Union 7th Framework Programme—FP7-HEALTH-2013-INNOVATION-2 (ASPRE Project # 601852) and the Fetal Medicine Foundation (FMF) (Charity No: 1037116).
Disclaimer The views expressed in this publication are those of the author(s) and not necessarily those of the FMF, European Union FP7, healthcare systems or competent authorities.
Competing interests None declared.
Ethics approval This study will be conducted in accordance with the principles of Good Clinical Practice. This protocol was submitted to the National Research Ethics Committee and a favourable ethical opinion was granted. The reference number is 13/LO/1479. Subsequent approval by individual ethical committee and competent authority was granted.
Provenance and peer review Not commissioned; externally peer reviewed.