Article Text

Abstract
Introduction Currently available analgesics are ineffective in 30–50% of patients suffering from neuropathic pain and often induce deleterious side effects. T-type calcium channel blockers (mibefradil, ethosuximide, NNC 55-0396) are of great interest for the development of new symptomatic treatments of neuropathic pain, due to their various effects on pain perception. Interestingly, ethosuximide, which has already been approved for treating epilepsy, is available on the European market for clinical use. Despite numerous preclinical data demonstrating an antinociceptive effect of ethosuximide in various animal models of neuropathic pain, no clinical studies have been published to date on the analgesic efficacy of ethosuximide in patients with neuropathic pain.
Methods and analysis The Ethosuximide in the Treatment of non-Diabetic Peripheral Neuropathic Pain (EDONOT) trial is a randomised, parallel, controlled, double-blinded, multicentre clinical study. It is the first clinical trial to evaluate the efficacy and safety of ethosuximide in the treatment of non-diabetic peripheral neuropathic pain. Adult patients exhibiting peripheral neuropathic pain (Numeric Rating Scale (NRS) ≥4 and Douleur Neuropathique 4 (DN4)≥4) for at least 3 months and under stable analgesic treatment for at least 1 month will be included. Patients (n=220) will be randomly assigned to receive either ethosuximide or control treatment for 6 weeks following a 1 week run-in period. The primary end point is the intensity of neuropathic pain, assessed by NRS (0–10) before and after 6 weeks of treatment. The secondary end points are safety (adverse events are collected during the study: daily by the patient on the logbook and during planned phone calls by investigators), the intensity and features of neuropathic pain (assessed by Brief Pain Inventory (BPI) and Neuropathic Pain Symptom Inventory (NPSI) questionnaires) and health-related quality of life (assessed by Medical Outcome Study Short Form 12 (MOS SF-12) and Leeds questionnaires).
Ethics and communication The study was approved by an independent ethics committee (CPP Sud-Est VI, France, IRB00008526) and registered by the French competent authority (Agence nationale de sécurité du médicament (ANSM)).
Trial registration number NCT02100046, Recruiting.
- PAIN MANAGEMENT
- ethosuximide
- T-type calcium channels
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Strengths and limitations of this study
Randomised, double-blinded and controlled study.
Larger study (220 patients).
The treatment duration is shorter (6 weeks) than the current recommendations (≥12 weeks).
An inactive control rather than a placebo.
Patients with diabetes were not considered.
Introduction
Currently, there is evidence that voltage-gated calcium channels modulate pain perception due to their influence on neuronal transmission and excitability. In the past, attention was focused on the modulation of high voltage-activated calcium channels (Cav1 and Cav2 families). Recently, scientific interest has turned to low voltage-activated calcium channels (Cav3 family), so-called T-type channels. Since this change of direction, the literature has emphasised significant involvement of these channels in the physiology of nociception and chronic pain processes (for a review see ref. 1).
The analgesics currently available often lack efficacy in the treatment of neuropathic pain and have fairly poor tolerability (for a review see ref. 2). Thus, the clinical use of inhibitors of T-type calcium channels would not only help the development of new therapies for the treatment of neuropathic pain, whose prevalence is estimated at 7–8% in Europe (5% for moderate and severe pain),3–9 but it would also have an economic impact due to the low sales price of the currently available inhibitor, Zarontin (200 mL syrup; 250 mg/5 mL).
T-type calcium channels and pain
Cloning the α-1 subunit of T-type channels revealed at least three subtypes: α-1G (Cav3.1),10 α-1H (Cav3.2)11 and α-1I (Cav3.3).12 T-type calcium channels possess a unique property in neuronal excitability processes:13 ,14 activation by weak depolarisation of the cell membrane. This makes them capable of playing a role in different neurophysiological processes in neurons: the initiation of action potentials or spike trains, intracellular calcium influx, the release of neurotransmitters and the amplification of weak dendritic signals (inhibitory and excitatory postsynaptic potentials; for a review see ref. 1).
Several in vitro and in vivo studies have identified various functions of T-type calcium channels, including their involvement in nociception and their contribution in the development of acute and chronic pain (neuropathic, visceral and inflammatory).15–19 T-type channel inhibition achieved by various experimental approaches (genetic and pharmacological) induces an analgesic effect in different types of painful conditions and in various pathological contexts. The main pharmacological compounds used to assess the analgesic properties of T-type channels are ethosuximide20 and mibefradil.21
Ethosuximide and peripheral neuropathic pain
Ethosuximide is currently used in children and adults to treat absence seizures but it has no indication for pain relief. Considering the role of T-type calcium channels in nociceptive processes, several preclinical studies have investigated the effect of ethosuximide on pain, especially in the context of neuropathic pain. According to these studies, ethosuximide exhibited a relevant analgesic and antihyperalgesic effect and reduced painful symptoms related to neuropathic syndrome, suggesting the role of T-type calcium channels in the initiation and maintenance of neuropathic pain.
Ethosuximide possesses a moderate analgesic property in healthy rats22 but completely removes painful neuropathic symptoms induced by sciatic and spinal nerve ligation.23
Several cytotoxic chemotherapies may induce neuropathic syndrome (including neuropathic pain), thus limiting their use in patients. Flatters and Bennett24 showed that systemic injection of ethosuximide relieved mechanical and thermal cold allodynia in rats after receiving anticancer therapy (paclitaxel or vincristine). Similar results were found in a model of oxaliplatin-induced peripheral neuropathy.25 In addition, the chronic use of ethosuximide in these animal models does not induce tolerance.
No preclinical studies have evaluated the potential benefit of the use of ethosuximide in the context of diabetic neuropathy, but two preclinical studies have shown a relationship between T-type channels (Cav3.2 member) and diabetic neuropathy: the knock-out animals for Cav3.2 channels26 and receiving Cav3.2 antisense RNA27 do not develop allodynia or hyperalgesia induced by diabetic neuropathy.
Ethosuximide, action mechanism and safety in humans
Ethosuximide is a succinimide anticonvulsant, which is marketed since 1988 and is on the WHO's List of Essential Medicines.28 It is considered as the first-line treatment for treatment of absence seizures, partly because it is devoid of idiosyncratic hepatotoxicity, contrary to valproic acid, the alternative treatment 29 The mechanism by which ethosuximide affects neuronal excitability mainly includes block of T-type calcium channels, and may include effects of the drug on other classes of ion channels.30–32
Ethosuximide is rapidly and almost completely absorbed after oral administration. Peak plasma levels of 38 µg/mL are observed on average 3–7 hours after administration of a single dose of 500 mg in children. In adults, steady state is reached in about 7 days. The residual plasma levels are at 34 µg/mL on average, for a daily intake of 500 mg. Therapeutic blood levels of ethosuximide vary between 40 and 100 µg/mL. The volume of distribution of ethosuximide is about 0.7 L/kg. It does not bind to plasma proteins. It is found in the cerebrospinal fluid, saliva, tears and breast milk in concentrations similar to those of the plasma. It is extensively metabolised mainly by oxidative pathway in at least three metabolites. Only 20% of the administered dose is recovered in urine. The primary metabolite in urine and representing 60% of the total dose is 2-(1-hydroxyethyl)-2-methyl suximide.
The half-life of plasma elimination is ∼60 hours in adults and about 30 hours in children due to a higher metabolic clearance.
The safety profile of ethosuximide is similar with other antiepileptic drugs (AEDs). Frequently listed side effects are dyspepsia (nausea, epigastric pain, bloating and loss of appetite), dizziness, headache, ataxia, skin rash and vomiting. The potential advantage of ethosuximide resides in its mode of action that is different from other AEDs used in neuropathic pain treatment. Indeed, gabapentinoids act primarily on the α-2 δ subunit of calcium channel,33 ,34 while ethosuximide acts primarily on T-type calcium channels (having no α-2 δ subunit).13 This specific pharmacological profile could provide ethosuximide same efficacy in patients who are not sufficiently relieved by gabapentinoids.
No clinical studies have evaluated the potential benefit of the use of ethosuximide in the context of peripheral neuropathy. If the analgesic efficacy and good safety of ethosuximide is demonstrated, a comparison with other reference AEDs in the treatment of neuropathic pain should be considered.
Rationale for this pilot study
Owing to their various involvements in the development and maintenance of chronic pain, T-type calcium channels are of great interest for the development of new symptomatic treatments of neuropathic pain. Analgesics available in our pharmacopoeia are ineffective and poorly tolerated in many patients with neuropathic pain. Interestingly, Zarontin which contains the active substance ethosuximide (T-type calcium blocker) is available on the European market for epilepsy. Despite this and numerous preclinical data demonstrating the antinociceptive effect of ethosuximide in various models of neuropathic pain, no clinical studies have been published to date on the therapeutic efficacy of ethosuximide in patients with neuropathic pain. Preclinical arguments and the absence of clinical evaluation provide the rationale for conducting the first pilot clinical trial to assess the potential benefit of using ethosuximide in the treatment of neuropathic pain.
Methods and analysis
The present study is a randomised, parallel, controlled, double-blinded and multicentre phase II clinical trial to evaluate the efficacy and safety of ethosuximide in patients with non-diabetic peripheral neuropathic pain. Two hundred and twenty patients from 19 clinical sites in France are planned for inclusion. The study duration for each patient included will be 7 weeks, including a 1 week run-in period and 6 weeks of treatment.
Study objectives
The primary objective of this study is to evaluate the analgesic efficacy of ethosuximide administered in addition to background therapy to patients with peripheral neuropathic pain, versus inactive control.
Secondary objectives will be to study the effects of ethosuximide on:
The intensity of daily pain (average and maximum pain experienced) throughout the study,
The characteristics of neuropathic pain at the end of 6 weeks treatment,
The health-related quality of life (HRQoL; physical and mental) at the end of 6 weeks treatment,
The patient's quality of sleep throughout the study,
The Patient's Global Impression of Change (PGIC) at the end of 6 weeks treatment.
Inclusion and exclusion criteria
Participants will be patients with peripheral neuropathic pain diagnosed for more than 3 months and not relieved by the usual treatments (see details in box 1).
Inclusion and exclusion criteria of the study
Inclusion criteria
Man or woman aged 18 or over.
Negative pregnancy test and effective contraception.
Peripheral neuropathic pain diagnosis ( Douleur Neuropathique 4 (DN4) ≥4).35
Treatment failure (Numeric Rating Scale (NRS) pain ≥4) for at least 3 months despite stable analgesic treatment for 1 month.
Normal liver function (Alanine Aminotransferase (ALT), Aspartate Aminotransferase (AST), Alkaline Phosphatase (ALP), Gamma-GT (GGT) <3N).
Normal renal function (creatininaemia <133 μmol/L).
Haematocrit >38% (men) and >34% (women).
Patients affiliated with the French Social Security system.
Patients able to provide free and informed consent.
Exclusion criteria
Breast feeding.
Central neuropathic pain (spinal or supraspinal), stroke type or spinal cord injury, phantom limb pain, fibromyalgia.
Medical and surgical history incompatible with the study.
Addiction to alcohol and/or drugs.
Taking antiepileptics (carboxamide family).
Patient treated with ethosuximide.
Allergy to succinimide (ethosuximide, methsuximide, phensuximide).
Psychotic disorders.
Epilepsy.
Malabsorption of glucose and galactose.
Patients with diabetes.
Sucrase/isomaltase deficit.
Participation in another clinical trial.
Clinical trial exclusion period.
Total amount of compensation higher than €4500 for the 12 months preceding the start of the trial.
Insufficient cooperation and understanding to adhere strictly to the conditions demanded by the study.
Patients subject to legal protection.
No therapeutic change will be generated by the protocol; patients will be treated with ethosuximide or inactive control in addition to their current treatment for neuropathic pain. However, no therapeutic change for neuropathic pain will be allowed at any time during the study.
Patients can be withdrawn from the study for any of the following reasons: modification of the analgesic therapy, intolerance to ethosuximide, withdrawal of consent, breach of protocol, significant adverse events.
Investigational medicinal product
To ensure the double-blind condition, therapeutic units will be kept in similar bottles and labelling will be performed to mask brand names.
Zarontin (Pfizer, ethosuximide)
The active substance is ethosuximide. This is an AED and a T-type calcium channel blocker. It is currently authorised in Europe for the treatment of epilepsy. It is active on absence seizures and used alone or in combination with another AED in the treatment of generalised epilepsies.
Ethosuximide will be administered two times a day, morning and evening during meals for 42 days (6 weeks). The dosage will be increased very gradually 250 mg (5 mL) every 4 days until reaching the maximum dose at 20 mg/kg (or 1500 mg) per day, which corresponds to the current summary of product characteristics. However, if during the titration phase, the patient reports uncomfortable adverse effects, the investigator has the option to continue treatment at the lower dose level, if well tolerated, up to the end of the study. This pragmatic attitude corresponds to current clinical practice and is aimed at reducing the risk of study discontinuation. It will also provide information on the dose with the best benefit/risk ratio in this indication.
Inactive control
Stodal (Boiron Laboratories) is a homeopathic syrup indicated for the treatment of cough. The choice of this homeopathic syrup as inactive control is justified by several arguments:
The syrup bottle packaging of Stodal is similar to that of Zarontin: pharmaceutical form (syrup), bottle shape, colour and volume. These similarities conform to the double-blind requirements.
It is a homeopathic treatment traditionally used in the treatment of cough, and has no indication in the treatment of pain.
According to the recommendations of the painkiller studies mentioned in the Cochrane review of Moore et al36 dealing with clinical studies evaluating the efficacy of analgesics on neuropathic pain, and according to good clinical practice, it is recommended that the first phase II study should evaluate the efficacy of analgesics tested in comparison with a placebo or inactive control. In addition, current active comparators, namely pregabalin and gabapentin, are marketed in capsule or tablet form, which would require implementing a double dummy.
Stodal will be taken for 42 days (6 weeks) with the same administration modalities as the ethosuximide group.
Study end points
Study end points were based on the recommendation of the European Medicinal Agency (EMA; International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) guideline on neuropathic pain: Committee for Proprietary Medicinal Product (CPMP)/Efficacy Working Party (EWP)/252/03 Rev. 1).
Primary end point
Pain intensity (NRS, 11 points): This numeric scale allows the patient to rate their pain from 0 to 10, with 0 for no pain and 10 for the worst possible pain. Average pain intensity over the past 24 hours is recorded once daily, before bedtime, on the logbook throughout the study and the values are averaged for the 7 days preceding the two time points D0 (baseline) and D0+42 (last visit). The primary end point is the change in Numeric Rating Scale (NRS) at D0 and D0+42.
Secondary end points
Health-related quality of life (MOS SF-12): HRQoL will be evaluated by the MOS SF-1237 questionnaire which assesses the physical and mental health of the patient using 12 questions related to eight aspects of health (physical and social activities, morale, physical and emotional strength to accomplish everyday tasks, physical pain, general mental health, vitality, perceived general health status). A score is determined for physical and mental health (0–100).
Neuropathic pain symptom (NPSI and BPI): The Neuropathic Pain Symptom Inventory (NPSI)38 and the Brief Pain Inventory (BPI)39 will be used to evaluate the characteristics and impacts of neuropathic pain.
The NPSI is a self-administered questionnaire designed to assess different symptoms of neuropathic pain. It includes 12 items that can discriminate and quantify five separate clinically relevant dimensions.
The BPI is a self-administered questionnaire which includes: (1) a body scheme, (2) the maximum pain, less pain, usual pain during the past 15 days (NRS 11 points), (3) the description of the analgesic treatment in progress, (4) an evaluation of pain relief on a percentage scale (0–100%), (5) the impact of pain on mood, relationships with other people, walking, sleeping, work, joy of life, leisure activities (NRS, 0 normal to 10 impossible).
Quality of sleep (LSEQ): The Leeds Sleep Evaluation Questionnaire (LSEQ)40 is a standardised questionnaire composed of 10 self-visual analogue scales (10 cm) that relate four aspects of sleep efficiency: (1) the quality of falling asleep and level of sleepiness, (2) sleep quality, (3) awakening quality, and (4) the quality of state after awakening and performance.
These evaluations will be conducted during the screening visit (D0) and at the end of the study (D0+42).
Moreover, daily evaluation (in the morning) of the quality of falling asleep and sleep during the previous night (NRS from 0 (very poor) to 10 (excellent)) will be reported on the daily logbook.
Patient's Global Impression of Change: PGIC41 is aimed at assessing the general effectiveness of the treatment. This scale consists of seven level descriptors answering the question ‘How are you?’ distributed in three ways: (1) improved (very/medium/slightly), (2) unchanged and (3) aggravated (slight/medium/very).
Safety: Any adverse events were collected daily by the patient and during the planned phone calls by investigators (every 4 days). Adverse events are categorised according to their type, intensity and treatment related by investigators. Owing to the long half-life of ethosuximide (60 hours), a phone call, within 2 weeks after stopping treatment, will be allowed to recover any adverse effects.
Methodology and study design
The study methodology was selected on the recommendation of the EMA (ICH guideline neuropathic pain: CPMP/EWP/252/03 Rev. 1).
Patients will be treated for 6 weeks either by ethosuximide, according to a scheme of specific titration (maximum dose of 1500 mg/day achieved in 20 days) or an inactive control treatment administered with the same modalities (see figure 1).
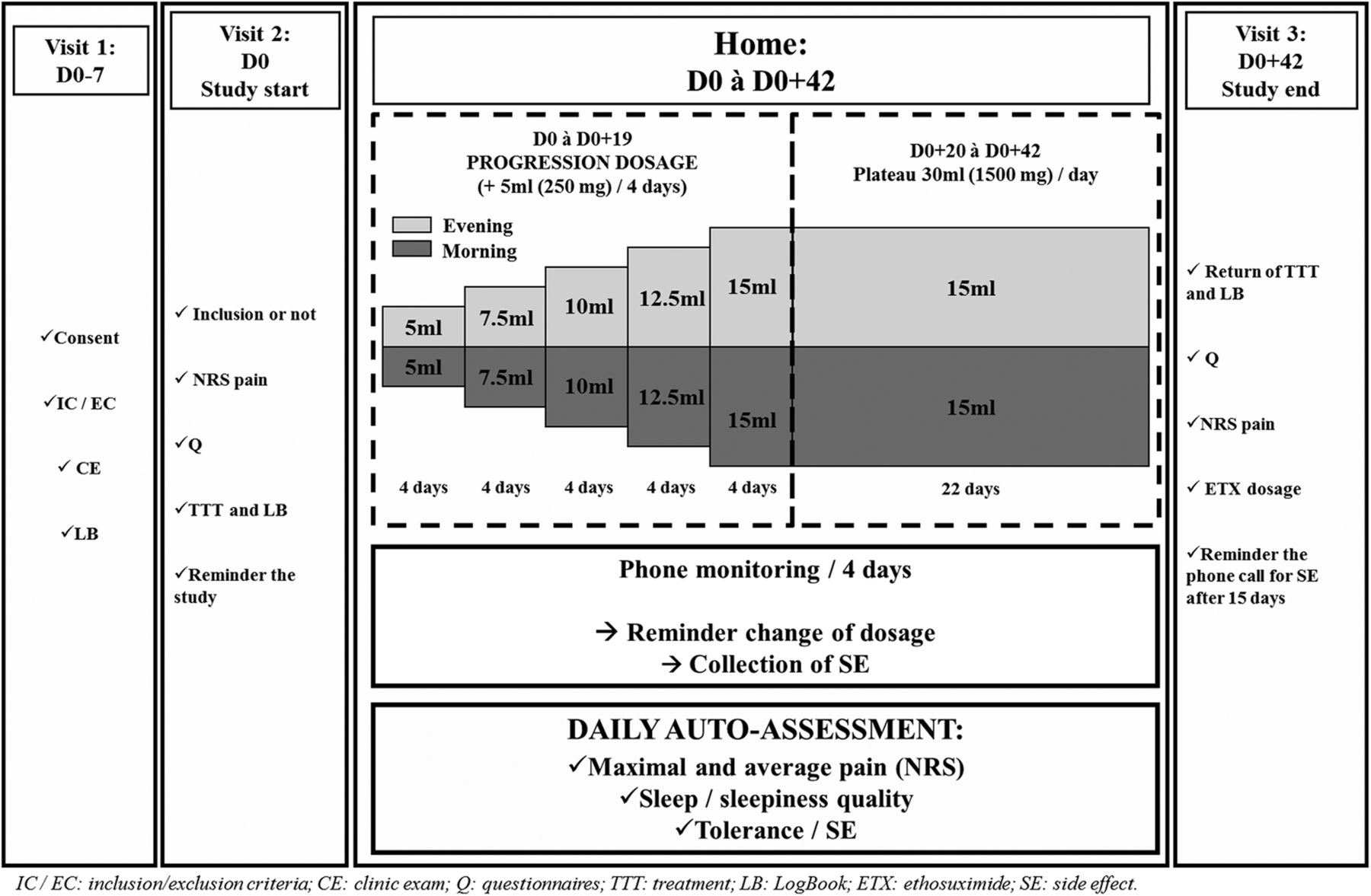
{kind=link}
Study design. NRS, Numeric Rating Scale; IC, Inclusion criteria; EC, Exclusion criteria; CE, Clinic exam; LB, Logbook; Q, Questionnaire; TTT, Treatment; SE, Side effect; ETX, Ethosuximide.
Enrolment
Patients followed up in their referral centre for the treatment of pain will be preselected. Patients will be contacted in order to briefly present the purpose of the study and make an appointment for the inclusion visit.
Visit 1—inclusion (D-7) and run-in period (D-7 to D0)
The objectives of the study, practice organisation, constraints and different questionnaires will be explained in detail to the patient by the investigator who will also collect the informed consent form. The patient must present a neuropathic pain diagnostic defined according to the DN4 questionnaire and the International Association for the Study of Pain (IASP) criteria determined during the clinical examination by the investigating physician.
A daily logbook will be given to the patient along with detailed instructions to collect every day, the median and maximum pain score experienced during the day and sleep and falling asleep quality. Possible side effects should be collected. The patient will return home with a logbook for 7 days, corresponding to the run-in period assessing the patient's ability to complete the logbook and recover the data on the intensity of neuropathic pain.
This run-in period, without any studied treatment, was introduced (1) to evaluate the ability of the patients to rate their pain daily in their logbook; (2) to check the basal average pain intensity over a 1-week period. The patients received the treatment after this run-in period only if the inclusion criteria were met (NRS pain ≥4 and completed logbook as required by the protocol).
Visit 2—start of treatment (D0)
Daily average pain scores have to be filled-in by the patient on the logbook during the past 7 days and the average pain score should be ≥4 to include the patient in the study. The patients have to fill in four questionnaires: (1) LSEQ, (2) MOS SF-12, (3) NPSI and (4) BPI.
If all the inclusion and non-inclusion criteria are conformed to, the patient will be enrolled in the study and randomised in one of the two treatment arms (ethosuximide or inactive control). The administration dosages, according to a specific titration scheme, will be explained in detail to the patient. At the end of the visit, the patient will receive all the therapeutic units for the duration of administration required by the study protocol (42 days).
Ambulatory period (D0 to D0+42)
Ethosuximide or inactive control treatment will be administered in two daily doses during meals, according to a scheme of specific titration for 20 days followed by a plateau at the maximum dose of 1500 mg/day for 22 days. The patient will assess and record the following daily in the logbook: the quality of falling asleep and sleep during the previous night and the median and maximum pain felt in the day.
Every 4 days (at the end of each dose escalation level), patients will be contacted by phone in order to collect information on any side effects.
Visit 3—study end (D0+42)
Idem visit 2. The patient must also complete the PGIC questionnaire.
This visit represents the end of study.
Owing to the long half-life of ethosuximide (60 hours), telephone contact, within 2 weeks after stopping treatment, will be allowed to recover from any adverse effects.
Statistical considerations
Sample size estimation
According to previous works,36 ,42 pain intensity was estimated at around 2.5. Hundred patients per group will be included to highlight a difference equals to 1 for a two-sided type I error at 5% and a statistical power at 80%. Finally, a total of N=220 patients will be considered to take into account lost to follow-up (10%). An interim analysis is planned after enrolment of the first 110 patients using the Lan and DeMets rule (Pocock, East software, Cytel, Cambridge, Massachusetts, USA). The type I error is fixed at 0.003 for this interim analysis.
Statistical analysis
Statistical analyses will be conducted using Stata software (V.13, StataCorp, College Station, Texas, USA). A two-sided p value of <0.05 will be considered to indicate statistical significance (except interim analysis). Comparisons between independent groups will be analysed using the χ2 or Fisher's exact test for categorical variables (notably PGIC and safety) and Student's t-test or Mann-Whitney's test for quantitative parameters (notably pain intensity, health-related quality of life scores measured by MOS SF-12, NPSI and BPI scores, sleep quality evaluated using Leeds score). Normality will be studied by the Shapiro-Wilk test and homoscedasticity using the Fisher-Snedecor test. Intention-to-treat will be considered for the primary analysis. The analysis of the primary outcome will be completed by multivariate analysis using a linear mixed model to take into account: (1) fixed-effects covariates chosen according to univariate results and to clinical relevance, and (2) centre as random-effects (to measure between-centre and within-centre variability). The two treatment groups will be compared for end of treatment data. Also included will be the analysis of repeated measures and random-effect models (linear or generalised linear). Other treatments will be considered as a covariate to study the impact on patient quality of life, pain intensity and quality of sleep. According to clinical relevance and to EMA and CONSORT recommendations, subgroup analysis depending on the aetiology (diabetic neuropathy, postherpetic, etc) will be proposed after the study of aetiology×randomisation group interaction in regression models (for repeated data or not). Finally, particular focus will be placed on lost to follow-up. A study of abandonment considered as censored data will be proposed using the Kaplan-Meier estimation. A sensitivity analysis will be performed and the nature of missing data will be studied (missing at random or not). According to this, the most appropriate approach to the imputation of missing data will be proposed (maximum bias (eg, last observation carried forward vs baseline observation carried forward) or estimation proposed by Verbeke and Molenberghs for repeated data).
Dissemination
Approval
Any substantial modification of the protocol and of the informed consent form will be presented to the independent Medical Ethics Committee. The latter and the competent French authority will be informed of the end of the study. In accordance with the independent Medical Ethics Committee (CPP Sud Est VI, Clermont-Ferrand, France), no safety and data monitoring committee has been set up in view of the low risk of the ethosuximide treatment. The study is currently registered on the clinical trials website under the following number: NCT02100046. The protocol has been in V.12 since 16/12/2015.
Patient informed consent
According to the French law on biomedical research, written informed consent must be obtained from patients prior to participation in the study. Patients will voluntarily confirm their willingness to participate in the study, after having been informed (in writing and verbally) by investigators of all aspects of the study that are relevant to their decision to participate. They will be informed about requirements concerning data protection and have to agree to direct access to their individual data. The patients will be informed that they are free to withdraw from the study at any time at their own discretion without necessarily giving reasons.
Data collection and quality management
A clinical research assistant will be dedicated to data entry, coding, security and storage. Each patient included and the study data will be anonymised. The study data will be collected and managed using a case report form. A clinical research assistant will be commissioned by the sponsor (University Hospital of Clermont-Ferrand) in order to monitor the progress of the study in accordance with the Standard Operating Procedures implemented in the University Hospital of Clermont-Ferrand, in accordance with Good Clinical Practices and current French laws.
Access to data and communication of results
The data set will be the property of the sponsor (University Hospital of Clermont-Ferrand). However, the principal investigator (AE) and the project manager (NK) will have full access to the final data set. The results will be communicated in a peer-reviewed journal, presented at international congresses and completed online on ClinicalTrials.gov.
Discussion
This translational research project aims to demonstrate the therapeutic effect of ethosuximide on peripheral neuropathic pain.
In this study, particular emphasis has been placed on the methodology in order to provide a sufficient level of evidence. Indeed, we relied on the recommendation of the EMA (ICH guideline on neuropathic pain: CPMP/EWP/252/03 Rev. 1) and endeavoured to comply with them as much as possible: randomised; controlled; double-blinded; parallel groups; large patient sample (110/arm); several weeks of treatment; mixed population and relevant evaluation criteria.
However, the present protocol has some limitations:
A 6-weeks treatment period was chosen, considering that this study is a proof-of-concept aimed at providing a first look on the efficacy and the safety of ethosuximide in patients with neuropathic pain. In the case of positive efficacy/tolerability ratio, a larger study with longer treatment duration (≥12 weeks) and an active comparator (ie, gabapentinoid) will be undertaken.
In this study, we used a homeopathic syrup (Stodal) as an inactive control. This homeopathic cough syrup may taste different than the study treatment. To lower the risk of bias as much as possible, the study design is in parallel groups and all patients included have to be naive for ethosuximide treatment. In addition, investigators are not in contact with the treatment, which is always dispensed by pharmacists.
This choice was made also for scientific motives. Indeed, several studies have shown that the effect of homeopathic treatment is not better than that of a placebo (for a review see ref. 43). Owing to conflicting studies,44 this choice was made assuming that if homeopathy does have a therapeutic effect, and that ethosuximide has a greater effect than our inactive control group, then this will give even greater credence to the therapeutic effect observed. Moreover, no data were found when searching for links between the terms ‘Stodal’ and all these components and ‘pain’ on MEDLINE. This provides further evidence of the lack of analgesic effect of Stodal and these components.
Neuropathic pain remains an important public health issue, as its prevalence in Europe is estimated at 7–8%3–9 and up to 40% of the patients treated are not or poorly relieved by current treatments (for review see refs. 45 and 46). Peripheral neuropathy symptoms can last for months and even years after surgery, chemotherapy, herpetic infection, diabetes, trauma, etc, and the lack of support induces anxiety, depression, sleep disorders and a decrease of HRQoL (for reviews see refs. 47–49). Neuropathic pain has a strong economic and social impact, as patients with neuropathic pain generate significant excess healthcare costs and resource use amounting to €10 313 per patient per year in France.50 Therefore, innovative therapeutic strategies are now more than necessary to treat neuropathic pain. This is the first study to examine the effectiveness of ethosuximide on neuropathic pain. If the results are positive, it will be an important step forward for the pharmacopoeia of this pathology.
References
Footnotes
Contributors NK, AE, CC, CM, ChD and ClD led and contributed to the conceptualisation, design and implementation of this research protocol. BP led the development of the statistical analysis plan. NK participated in the design of the protocol for interventions and assessments. All the authors have read and approved the final manuscript.
Funding This work is supported by funding from the French Ministry of Health (Programme Hospitalier de Recherche Clinique Interrégional, year 2013) and the APICIL Foundation.
Competing interests None declared.
Patient consent Obtained.
Ethics approval The approval of the Medical Ethics Committee (CPP Sud Est VI, Clermont-Ferrand, France) was obtained on 14 February 2014. The protocol was declared to the competent French authority (Agence Nationale de Sécurité du Médicament et des produits de santé) and registered under the number 131567A-32. Authorisation was obtained on 26 March 2014.
Provenance and peer review Not commissioned; externally peer reviewed.