Article Text

Abstract
Introduction Perioperative chemotherapy is the gold standard treatment of the resectable gastro-oesophageal adenocarcinoma. However, 70% of patients cannot receive the complete sequence because of a postoperative complication or a decrease in functional and nutritional reserves. Recently, a new concept appeared in digestive surgery: prehabilitation. This interventional process consists of patient preparation, between surgical consultation and surgery, and is based on 3 components: (1) physical management, (2) nutritional care and (3) psychological care. Prehabilitation should decrease postoperative complications and improve nutritional and physical status during the preoperative and postoperative periods. Therefore, it is becoming essential to evaluate the effect of prehabilitation, compared to conventional care, on the percentage of patients reaching the complete oncological treatment.
Methods and analysis The PREHAB trial aimed to evaluate the efficacy of prehabilitation compared to conventional care, in patients with gastro-oesophageal cancer with perioperative chemotherapy. This trial is a prospective, randomised, controlled, open-blind and interventional study in 4 centres. Patients (n=60 per group) will be randomly assigned for management with either prehabilitation or conventional care. The primary outcome is the percentage of patients reaching the complete oncological treatment decided in a multidisciplinary tumour board. The secondary outcomes are the postoperative morbidity, disease-free survival, overall survival, feasibility of the protocol, length of stay, variation of the functional reserve after the preoperative chemotherapy (defined by the VO2peak, ventilatory threshold and 6-min walk test), preoperative and postoperative nutritional status, preoperative anxiety, quality of life, 30-day and 90-day mortality and cumulative dose of cytotoxic treatment received.
Ethics and dissemination The study was approved by an independent medical ethics committee (IRB00008526, CPP Sud-Est VI, Clermont-Ferrand, France) and by the competent French authority (ANSM, Saint Denis, France) and registered on Clinicaltrial.gov. The results will be disseminated in a peer-reviewed journal.
Trial registration number NCT02780921.
- Prehabilitation
- Gastric cancer
- Fitness
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Strengths and limitations of this study
A multicentric, prospective and randomised study.
Large number of participants (n=120).
Prehabilitation includes nutritional care and psychological treatment.
Intraoperative and postoperative protocols are not standardised.
This study includes oesophageal and stomach cancer.
Introduction
Perioperative chemotherapy is the gold standard treatment of the resectable and advanced gastro-oesophageal adenocarcinoma. The efficacy of this strategy has been demonstrated in two randomised studies.1 ,2 It reduces tumour size before surgery, treats micrometastases and evaluates chemosensitivity. Disease-free survival (DFS) and overall survival (OS) rates were significantly improved with perioperative chemotherapy compared to surgery alone. However, the limitation of these studies is that among all patients requiring chemotherapy, almost 70% of patients did not receive the complete sequence. This sequence is defined by the administration of two to four cycles before and two to four cycles after the surgery, according to the protocol. The major cause of absence or impossibility of realisation of postoperative chemotherapy was the presence of postoperative complication, postoperative serious asthenia and impaired nutritional and physical status.1 ,2 Poor physical condition assessed by cardiopulmonary exercise testing, reflecting a reduced physiological reserve, is predictive of postoperative complications.3 ,4 Physical training, even during a short period and on a various population, is beneficial in improving physical condition, cardiopulmonary function and muscular mass of the patient.5–8 Prehabilitation over a 6-week period between pre-surgical clinic appointment and surgery decreases postoperative morbidity and the hospital stay in cardiovascular surgery, but no study has ever been performed in patients presenting with gastric or oesophageal cancer.7 ,9–11
Prehabilitation revolves around three axes: (1) physical training based on initial cardiopulmonary exercise testing (VO2peak, ventilatory threshold (VT) and 6 min walk test (6MWT)), three times by week, supervised by a physical therapist; (2) nutritional care to ensure the compliance of the nutrition programme and adapt the nutritional management based on protein and energy needs and on the level of spontaneous oral intake; and (3) psychological treatment by a psychologist to reduce preoperative anxiety. To the best of our knowledge, no study has ever focused on gastro-oesophageal cancer. The benefit of prehabilitation in this cancer may be particularly important because (1) this surgery is associated with a high 90-day morbidity (40%, especially respiratory) and 90-day mortality (5%), (2) the physical and nutritional status of these patients is often precarious (cancer cachexia, gastro-oesophageal obstruction) and (3) the need to preoperative chemotherapy declines physical reserves and is associated with a lengthening of the time between presurgery clinic appointment and surgery of more than 3 months.12 Also, we hypothesise that, in this parallel and non-inferiority study, a physical training, personalised nutritional support and psychologist global management would results in an increase of numbers of patients receiving their full cancer treatment, by a decrease of postoperative complications and an increase of postoperative nutritional status. The aim of this study was to compare the percentage of patients reaching the complete oncological treatment previously decided in a multidisciplinary tumour board in the group with prehabilitation to the group with conventional care, in patients with gastro-oesophageal adenocarcinoma.
Methods and analysis
Study setting
The present study is a prospective, randomised, controlled, open and multicentric phase III trial that compares prehabilitation (Prehab group) versus conventional care (control group) in patients presenting with gastric and low oesophageal adenocarcinoma, treated by perioperative chemotherapy. Inclusions will be performed in four French tertiary centres (figure 1).
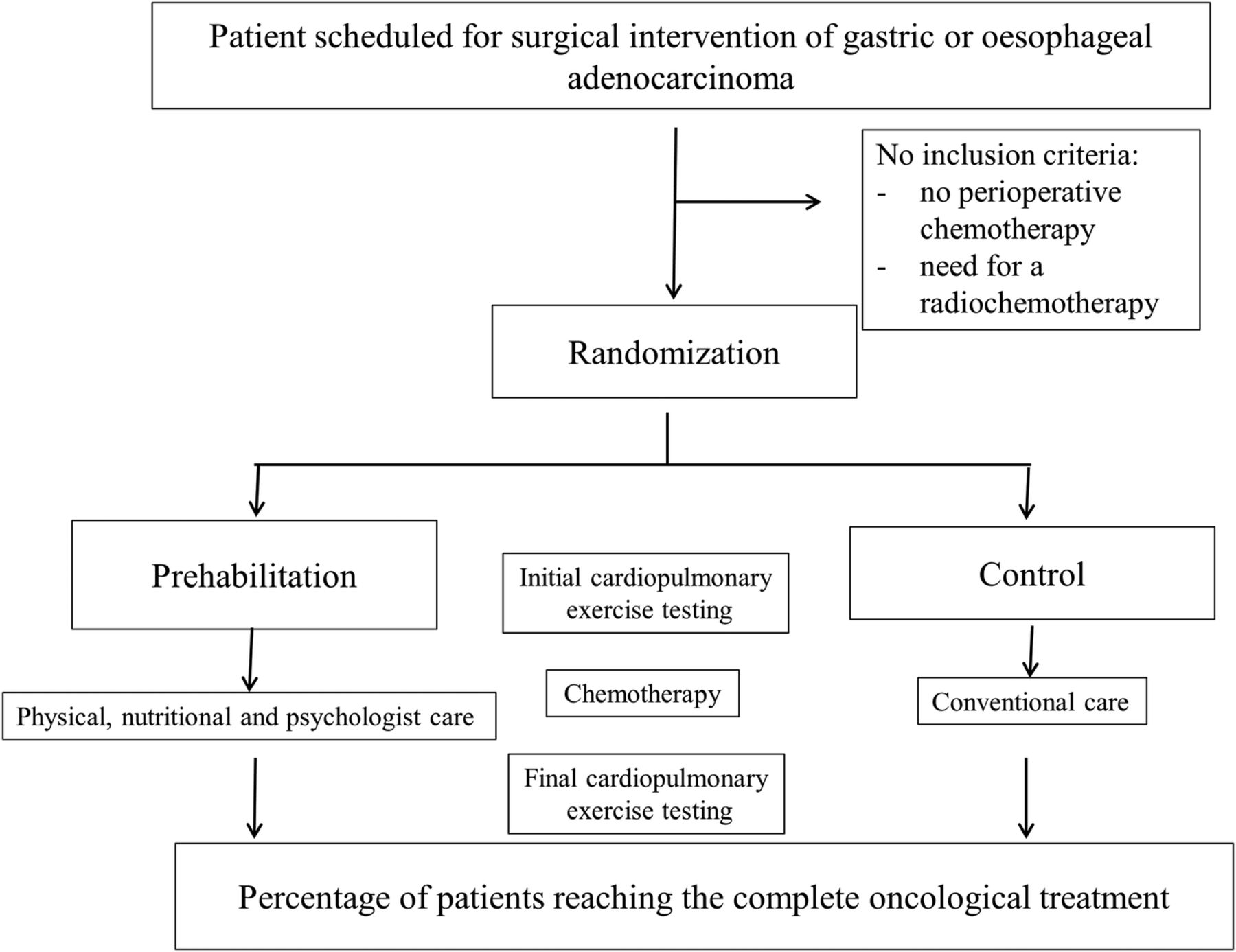
Consort diagram: flow chart.
Study objectives
In the experimental group (Prehab group), the main objective was to demonstrate an improvement in the percentage of patients reaching the complete oncological treatment fixed in a multidisciplinary tumour board. The secondary objectives was to evaluate the effect of the prehabilitation on the postoperative morbidity according to the Dindo-Clavien classification and Comprehensive Complication Index (CCI), severe morbidity (Clavien >2), DFS, OS, feasibility of the protocol (number of physical sessions realised on the 18 proposed), length of stay, variation of the functional reserve after the chemotherapy (defined by VO2peak, VT and 6MWT), preoperative and postoperative nutritional status, preoperative anxiety, quality of life (EQ-5D survey) at the end of the treatment, 30-day and 90-day mortality and cumulative dose of cytotoxic treatment received.13
Inclusion and exclusion criteria
To be included in the study, the participant is scheduled for surgical intervention of gastric or oesophageal adenocarcinoma and received perioperative chemotherapy, insured under the French social security system and have signed documents to mark their free, prior and informed consent. Patients cannot be included in the study for one of the following criteria: <18 years of age, need for radiochemotherapy, treated for another cancer within 5 years, except basal cell skin carcinoma or carcinoma in situ of the cervix, presenting with cognitive disorders or major disability making impossible to understand the study and sign the informed consent, or being breastfeeding or pregnant. Finally, patients already included in another clinical trial, or estimated by the investigator to not be able to be compliant with the criteria of the study, or with legal incapacity (person deprived of liberty or subject under guardianship) will not be enrolled in this study. Guidelines regarding stopping participation are: withdrawal of patient consent, non-compliance of the patient, adverse event and by decision of the investigator. In case of withdrawal from the study, the patient will be followed up and managed normally in the digestive surgery department. The exclusion period during which the patient cannot participate in another clinical trial is 15 days before inclusion and 0 days after the end of the study.
Interventions
After the first visit to his surgeon, the patient will be presented to the multidisciplinary tumour board to validate the inclusion criteria and to schedule the number of cycles of preoperative and postoperative chemotherapy. After this step, a second consultation with the surgeon will take place to verify all inclusion and exclusion criteria and perform the randomisation. For the two groups, an initial (before chemotherapy) and final (1-week before the surgery) evaluation will be performed. The evaluation includes an exercise capacity evaluation by (1) an incremental symptom-limited cardiopulmonary exercise test on a cycloergometer according to international recommendations in order to determine the VO2peak, and the ventilator threshold (VT) and (2) a 6 min walk distance (6MWT) performed according to the ATS recommendations.14 ,15 Moreover, the patients will be assessed for a nutritional evaluation (albumin), bioelectric impedance analysis, evaluation of physical activity and ingesta, as well as evaluation of the level of depressive symptoms and anxiety with the HADS survey and the quality of life (5Q-FD survey).
Study group
Prehab group
Exercise intervention: The total-body exercise will consist of up to 1 hour of supervised exercise for at least 3 days per week, for a total of 18 sessions, alternating between aerobic and resistance training (figure 2). Exercise intensity will be prescribed based on the target heart rate obtained at VT during the initial CPET. The participant will exercise in the presence of the physical therapist who will provide corrective feedback, if necessary.

{kind=link}
{kind=link}
Study diagram.
Nutrition intervention: Initially, a nutritionist will perform a medical examination running appropriate biological tests to evaluate the nutritional status and to provide individualised care to each patient. Individual protein requirements will be calculated as 1.2 g of protein per kilogram of body weight (adjusted body weight was used for obese patients), as per the European Society for Clinical Nutrition and Metabolism (ESPEN) guidelines regarding surgical patients.16 Patients will be asked to consume the protein supplement within 1 hour of their exercise regimen to capitalise on postexercise muscle protein synthesis.17 Then, a dietician will assess the compliance of the nutritional support at each cycle of chemotherapy and will adjust it if necessary. After the preoperative chemotherapy, a second evaluation by a nutritionist will be performed.
Psychologist intervention: Patients will receive up to a 1 hour visit with a trained psychologist who will provide techniques aiming to reducing anxiety, such as relaxation exercises based on imagery and visualisation, together with breathing exercises. Each patient will practise these exercises with the psychologist initially and at each cycle of chemotherapy and at home two to three times per week. Once performed, the exercises at home will be marked on diaries. The psychologist also provides suggestions on how to enhance and reinforce patients' motivation to comply with the exercise and nutritional aspects of the intervention.
Control group
The control group will be treated according to conventional care and will not receive any specific intervention before surgery except nutritional support and physiotherapy at the surgeon's discretion.
Study outcomes
The primary outcome is, in patients presenting with gastric or oesophageal adenocarcinoma, the percentage of patients in each group receiving the full perioperative oncological treatment, previously defined by a multidisciplinary tumour board. If a patient does not complete a chemotherapy course (=event), he will be considered as a subject who did not receive the full protocol (chemotherapy-surgery-chemotherapy). However, a decrease in the dose of chemotherapy or a stop of a component of chemotherapy will not be considered as an event.
The secondary outcomes are: postoperative morbidity at 3 months according to the Dindo-Clavien classification and Comprehensive Complication Index (CCI); severe morbidity at 3 months (Clavien >2); DFS, survival defined by the time, in years, before recurrence at 3 and 5 years after the end of the postoperative chemotherapy; OS, defined by the time, in of the OS at 3 and 5; feasibility of the protocol defined by the percentage of physical sessions realised on the 18 beforehand suggested in the preoperative period; length (in days) of postoperative stay; difference between the initial (before preoperative chemotherapy) and final (after preoperative chemotherapy) VO2 at the VT (mL/min/kg); difference between the initial and final values of the VO2peak (mL/min/kg); difference between the initial and final 6MWT (in metres); difference between the initial and final weights (in kg); difference between the initial and final albuminemia (in g/L); difference between the initial and final evaluation on the score of the HADS survey (Hospital anxiety and depression scale) to assess the anxiety and depression from a survey with 14 questions; the difference in the score between the initial evaluation and at 3 months after the surgery of the quality of life as defined by the EQ-5D survey; 30-day and 90-day mortality.
Methodology and study design
The trial will be performed in four centres. Patients will be recruited, treated and followed up at the digestive surgery department of the University Hospital of Clermont-Ferrand (France), Lille (France), Lyon (France) and Rennes (France). Then, the multidisciplinary tumour board will check all the inclusion and exclusion criteria, the PREHAB trial will be proposed by surgeons to patients with gastric or oesophageal adenocarcinoma and with concomitant perioperative chemotherapy. Patients will be informed of the trial protocol and, on acceptance, will be randomised in the ‘Prehab’ group or the control group by the surgeon or the oncologist. Randomisation will be carried out using a dedicated centralised telephone system and accessible round the clock. The randomisation sequence will be generated by a biostatistician using random blocks and stratification as a function of the centres and type of cancer (œsophagus or stomach). The trial will be open blinded because of the procedures employed and with an objective primary end point. The patient will be informed of the randomisation arm throughout the trial. The only criteria, which will be recorded, to discontinue or, or modifying allocated interventions for a given trial participant is the participant request.
Statistical considerations
Estimation of sample size
According to previous works, we estimated the percentage of patients in each group, realising the full perioperative oncological treatment around 30%.1 ,2 A sample size of 56 patients by randomised group would provide 90% statistical power to detect an absolute difference of 30% (30% vs 60%) for a two-sided α level of 0.05. Finally, a total of 60 patients by group will be considered. An interim analysis is planned after enrolment of the first 60 patients using the Lan and DeMets, O'Brien-Fleming method (East software, Cytel, Cambridge, Massachusetts, USA). The type I error is fixed at 0.003 for this interim analysis. The time schedule of enrolment has been estimated at 18 months.
Statistical analysis
Statistical analysis will be conducted on intention to treat (ITT) using the Stata software, V.13 (StataCorp, College Station, TX, USA). A two-sided p value of <0.05 will be considered to indicate statistical significance (except interim analysis). Baseline characteristics will be presented for each randomised group as the mean±SD or the median (IQR) according to the statistical distribution for continuous data, and as the number of patients and associated percentages for categorical parameters. Comparisons between independent groups will be analysed using the χ2 or Fisher’s exact test for categorical variables (notably unplanned readmission and primary outcome: percentage of patients realising the full perioperative oncological treatment) and Student's t-test or Mann-Whitney’s test for quantitative parameters (notably weight loss, body mass index, albumin, body impedance, length of stay, VO2peak, VT, 6MWT, depressive and anxious symptoms evaluated using HADS, quality of Life according to EQ-5D). Normality will be studied by the Shapiro-Wilk test and homoscedasticity using the Fisher-Snedecor test. The analysis of the primary outcome will be complemented by multivariate analysis using the generalised linear mixed model (logistic for a dichotomous dependent variable) to take into account (1) fixed effects covariates retained according to univariate analysis results and clinical relevance and (2) random effects (between and within centre and surgeon variabilities). Censored data such as OS or event-free survival will be estimated using the Kaplan-Meier method and compared (1) by log-rank test in univariate situation and (2) using the Cox proportional hazard model in the multivariate context. Regarding the analysis of repeated-measures, random-effect models (linear or generalised linear) will be considered to study the fixed-effects group, time-points evaluation and interaction ‘group× time’, taking into account between-participant and within-participant variability.
In the prehab group, a dose–response study will be proposed to assess (1) the impact of the number of prehabilitation sessions really realised and (2) the compliance prehabilitation care (dietary and nutritional management). Particular focus will be placed on loss to follow-up. A study with the abandonment considered as censored data will be proposed using the Kaplan-Meier estimation. If the frequency of missing data is >5%, we will perform additional analyses using imputation methods.
Ethics and dissemination
Approval
In accordance with the Declaration of Helsinki and French regulations on clinical trials, the study was presented to an independent ethics committee, the ‘Comité de Protection des Personnes Sud Est 6’ (reference: AU1228, IRB00008526, Clermont-Ferrand, France). The approval of the committee was obtained on 7 March 2016. The protocol was declared to the competent French authority (“Agence Nationale de Sécurité du Médicament et des produits de santé”, Saint Denis, France) and registered under number 2015 A01733-46. Authorisation was obtained on 21 December 2015. Any substantial change in the protocol or in the informed consent form will be presented to both authorities as well as first inclusion, interim analyses and end of study. Data monitoring will be performed as per French regulations requirements. As required by the IRB, a Safety Monitoring Committee (DSMC) has been set up. Data will be collected at each trial visit (every 2 months) and at the interim analyses, regarding any adverse events (AE) and serious AE. All serious AE causally related to treatment procedures will be reported to the relevant ethics committees, the lead site and the independent data and DSMC for their review and recommendations. The DSMC comprises independent clinicians with an interest in prehabilitation and a statistician. Overview is carried out through the review of AE and serious AE, all of which are reported at the regular committee meetings. Each meeting determines the Board’s recommendation to the Steering Committee as to whether the study is safe to continue. The study is currently registered on the clinical trials website under the number NCT02780921. The current protocol version is the first since 20 April 2016.
Patient informed consent
According to international regulations on clinical trials, written informed consent will be obtained from patients prior to their participation in the study (see online supplementary appendices 1 and 2). Patients will voluntarily confirm their understanding and willingness to participate in the study after having been informed (in writing and verbally) by oncologists on all aspects of the study. They will also be informed about requirements regarding data protection and direct access to their individual data. The patients will be informed that they are free to withdraw from the study at any time at their own discretion, without necessarily giving reasons.
supplementary data
Data collection and quality management
Experienced and trained study coordinators will be dedicated to data acquisition, coding, security and storage, under the responsibility of investigators. Each study data will be anonymised. Data will be recorded in paper case report forms at the time of each patient contact. These will be faxed to the study lead site for checking followed by entry into the secure study database. Data will be collected and managed using REDCap electronic data capture tools hosted at the University Hospital of Clermont-Ferrand.18 Research Electronic Data Capture (REDCap) is a secure, web-based application designed to support data capture for research studies, providing: (1) an intuitive interface for validated data entry; (2) audit trails for tracking data handling and export procedures; (3) automated export procedures for seamless data downloads to common statistical packages; and (4) procedures for importing data from external sources. A clinical research assistant will be commissioned by the sponsor (University Hospital of Clermont-Ferrand) in order to monitor the progress of the study in accordance with the Standard Operating Procedures implemented at the University Hospital of Clermont-Ferrand, in accordance with the Good Clinical Practice and current French laws.
Access to data and dissemination of results
The data set will be the property of the sponsor (CHU Clermont-Ferrand). However, the principal investigator and the project manager will have full access to the final data set. The results will be communicated in a peer-reviewed journal, presented at international congresses and summarised on ClinicalTrials.gov.
Discussion
Perioperative chemotherapy became the gold standard treatment in advanced gastric and low oesophageal adenocarcinoma, with an improvement of DFS and OS.1 ,2 However, the limitation of these studies is that, among all patients requiring chemotherapy, 70% of patients will not receive the complete treatment sequence. In these studies, only patients in good nutritional and physical status without postoperative complications can receive postoperative treatment.1 ,2 A meta-analysis reported that prehabilitation improved postoperative morbidity, length of stay, nutritional and physical status.7 The PREHAB study presented here should demonstrate whether prehabilitation increases the percentage of patients who can receive the complete oncological treatment previously defined in a multidisciplinary tumour board, to increase DFS and OS.
References
Footnotes
Contributors BL, CB, FC, RR, BM, J-YM, MS, CP, JG, EF, KS, CM and DP led the conceptualisation, design and implementation of this research protocol with the collaboration of the FRENCH (Fédération de recherche en chirurgie digestive). BP led the development of the statistical analysis plan. BL participated in the design of the protocol for interventions and assessments. All authors have read and approved the final manuscript. BL, CB, FC, BP, CM and DP will have access to the final trial data set, ancillary study, posttrial care and disclosure of contractual agreements.
Funding This study has received a grant from PHRC (Protocole Hospitalier en Recherche Clinique) 2015 (PHRC IR 2015 LE ROY), a national public funding of research. In accordance with the Declaration of Helsinki and French regulations on clinical trials, the study was presented to an independent ethics committee, the ‘Comité de Protection des Personnes Sud Est 6’ (reference: AU1228, IRB00008526, Clermont-Ferrand, France). The approval of the committee was obtained on 7 March 2016. The protocol was declared to the competent French authority (“Agence Nationale de Sécurité du Médicament et des produits de santé”, Saint Denis, France) and registered under number 2015 A01733–46. Authorisation was obtained on 21 December 2015. A clinical research assistant will be commissioned by the sponsor (University Hospital of Clermont-Ferrand) in order to monitor the progress of the study in accordance with the Standard Operating Procedures implemented at the University Hospital of Clermont-Ferrand, in accordance with the Good Clinical Practice and current French laws. The data set will be the property of the sponsor (CHU Clermont-Ferrand). However, the principal investigator and the project manager will have full access to the final data set. The results will be communicated in a peer-reviewed journal, presented at international congresses and summarised on ClinicalTrials.gov. The study sponsor and funders had no role and will not have any authority over: study design; collection, management, analysis and interpretation of data; writing of the report; and the decision to submit the report for publication. Any substantial change in the protocol or in the informed consent form will be presented to the competent French authority (“Agence Nationale de Sécurité du Médicament et des produits de santé, Saint Denis, France) and a safety monitoring committee as well as first inclusion, the interim analyses and end of study.
Competing interests None declared.
Patient consent Obtained.
Ethics approval IRB00008526 and ANSM (2015-A01733-46).
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement The additional unpublished data can be available by the mail of the corresponding author.