Article Text

Abstract
Introduction Patients with atrial fibrillation frequently suffer from heart failure with preserved ejection fraction. At present there is no proven therapy to improve physical capacity and quality of life in participants with permanent atrial fibrillation with preserved left ventricular contractility.
Objective The single-centre IMproved exercise tolerance In heart failure With PReserved Ejection fraction by Spironolactone On myocardial fibrosiS In Atrial Fibrillation (IMPRESS-AF) trial aims to establish whether treatment with spironolactone as compared with placebo improves exercise tolerance (cardiopulmonary exercise testing), quality of life and diastolic function in patients with permanent atrial fibrillation.
Methods and analysis A total of 250 patients have been randomised in this double-blinded trial for 2-year treatment with 25 mg daily dose of spironolactone or matched placebo. Included participants are 50 years old or older, have permanent atrial fibrillation and ejection fraction >55%. Exclusion criteria include contraindications to spironolactone, poorly controlled hypertension and presence of severe comorbidities with life expectancy <2 years. The primary outcome is improvement in exercise tolerance at 2 years and key secondary outcomes include quality of life (assessed using the EuroQol EQ-5D-5L (EQ-5D) and Minnesota Living with Heart Failure (MLWHF) questionnaires), diastolic function and all-cause hospitalisation.
Ethics and dissemination The study has been approved by the National Research and Ethics Committee West Midlands—Coventry and Warwickshire (REC reference number 14/WM/1211). The results of the trial will be published in an international peer-reviewed journal.
Trial registration numbers EudraCT2014-003702-33; NCT02673463; Pre-results.
- atrial fibrillation
- heart failure with preserved ejection fraction
- spironolactone
- exercise tolerance
This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: http://creativecommons.org/licenses/by/4.0/
Statistics from Altmetric.com
Strengths and limitations of this study
Double-blinded randomised placebo-controlled study design.
Accurate assessment of exercise tolerance (the primary outcome) using cardiopulmonary exercise testing.
Recruitment from primary and secondary care settings to provide a representative population of patients.
Single-centre study.
Assessment of effect of the treatment for mortality is beyond the study statistical power.
Introduction
Heart failure (HF) with preserved ejection fraction (HFpEF) is an emerging problem of modern cardiology, represents about half of all cases of HF, and is very common in individuals with atrial fibrillation (AF).1–3 In the Framingham Heart Study, 37% of participants with new AF had HF and presence of AF was strongly related to incident HFpEF (HR 2.34, 95% CI 1.48 to 3.70).4 Despite preservation of left ventricular ejection fraction (LVEF), patients with HFpEF have poor quality of life, high morbidity and mortality; largely comparable to HF with reduced LVEF.5 Improvements in morbidity and mortality with conventional treatments used in HF with reduced LVEF, however, have not translated to HFpEF.6
AF is present in about 40% of participants with HFpEF and is associated with higher N-terminal pro b-type natriuretic peptide (NT-proBNP) levels, risk of death and hospital admission with HF.7–10 In the Candesartan in Heart failure-Assessment of Reduction in Mortality and morbidity (CHARM) programme, AF was associated with increased risk of death or hospitalisation for worsening HFpEF (HR 1.72, 95% CI 1.45 to 2.06 for adverse cardiovascular outcomes).8
The mechanisms leading to symptoms, morbidity and mortality in patients with HFpEF and AF are poorly understood. Under physiological conditions, left ventricular pressure rapidly decays after systole, allowing low filling pressures and adequate diastolic filling. In HFpEF, the diastolic filling is compromised as a result of aggravation in active and passive relaxation (increased cardiac stiffness).11 This ventricular filling abnormality, in turn, reduces cardiac output leads to symptoms of HF.1 This theory is supported by interventional experiments and by large population-based studies carried out using a non-invasive approach to measure diastolic stiffness.12–14 Furthermore, the elevated filling pressure will increase pressure in the pulmonary system and eventually lead to pulmonary hypertension and pulmonary oedema in acute settings. A stiff ventricle may possess only limited ability to use the Frank-Starling mechanism to increase stroke volume during exercise with increasing heart rates.15
While activation of profibrotic pathways is a known response to increased pressure load in the heart, increased production of myocardial collagen and development of fibrosis can also aggravate diastolic dysfunction and ventricular stiffness. Increased myocardial collagen turnover and shift in the balance between matrix metalloproteinases and their inhibitors also favour of excessive myocardial fibrosis.16 ,17
Aldosterone is an important promoter of left ventricular fibrosis.18 Mechanisms of aldosterone-mediated cardiac fibrosis include myocardial inflammation, oxidative stress, and cardiomyocyte apoptosis and also direct stimulation of cardiac fibroblasts to produce collagen.19 ,20 Cardiac expression of mineralocorticoid receptors is increased in AF, thus augmenting the genomic effects of aldosterone.21
The effectiveness of spironolactone in HFpEF has been tested recently in two clinical trials. The Aldosterone Receptor Blockade in Diastolic Heart Failure (ALDO-DHF) study mainly enrolled participants with hypertensive, another major risk factor for HFpEF.22 ,23 While 92% of the trial patients had hypertension, only 5% of the study population (n=22) had AF at presentation.22 ,23 The Treatment of Preserved Cardiac Function Heart Failure With an Aldosterone Antagonist (TOPCAT) study24 ,25 included a higher proportion of participants with AF (mainly paroxysmal AF). The study defined preserved left ventricular function as LVEF≥45%, thus recruiting a proportion of participants with impaired LVEF according to contemporary definitions (also called ‘HF with intermediate ejection fraction’ by some).1 ,26 Thus, the current evidence on the effectiveness of spironolactone in patients with AF with preserved LVEF on morbidity and quality of life is sparse. We, therefore, plan the IMproved exercise tolerance In heart failure With PReserved Ejection fraction by Spironolactone On myocardial fibrosiS In Atrial Fibrillation (IMPRESS-AF) trial to determine the effects of spironolactone in permanent AF with preserved LVEF.
Study objectives
The IMPRESS-AF trial aims to establish whether, in participants with permanent AF, treatment with spironolactone as compared with placebo will improve exercise tolerance as a surrogate for cardiovascular mortality/morbidity (primary outcome); and will improve quality of life and diastolic function, as well as reduce the rate of all-cause hospital admissions, and increase rate of spontaneous cardioversion to sinus rhythm (secondary outcomes). The IMPRESS-AF trial will provide evidence on the clinical effectiveness of a readily available treatment in participants with AF with preserved LVEF.
Study design
The IMPRESS-AF is a double-blinded, randomised, placebo-controlled single-centre trial conducted in Birmingham, UK. The trial aims to recruit 250 participants permanent AF and LVEF>55% from primary and secondary care to be randomised to either spironolactone or placebo. Recruitment of the planned 250 patients was completed on 29 June 2016. The trial protocol was developed following the Standard Protocol Items for Randomized Trials (SPIRIT) statement and the latest patient-reported outcome (PRO)-specific guidance from the International Society for Quality of Life Research (ISOQOL) Best Practice for PROs in trials taskforce.27–29 The full protocol is available (see online supplementary appendix 1).
supplementary appendix
Eligibility
The main inclusion and exclusion criteria are summarised in table 1. Eligible patients are of male or female gender and age of 50 years or older. Permanent AF is defined by the European Society of Cardiology criteria.30 ,31 All participants have LVEF>55% as established by echocardiography during the screening.32 The prospective participants must be able to perform cardiopulmonary exercise testing using a cycling ergometer and complete quality of life questionnaires in English in their native language. For this, an interpreter and translated materials are provided if English is not their spoken language. Average values from 10 consecutive cardiac cycles are calculated to establish LVEF and ratio of peak velocities of early diastolic mitral inflow and peak early tissue Doppler velocity (E/e’). In patients with hypertension, antihypertensive treatment was established before the recruitment and patients with systolic blood pressure more than 160 mm Hg were excluded.
Key eligibility criteria for IMPRESS-AF
To improve generalisability, we do not include a requirement for evidence of diastolic dysfunction, as the trial patients would have impaired diastolic function due to AF. The principal exclusion criteria are designed to exclude patients with contraindications to spironolactone or significant comorbidities, which would prevent the prospective participants from completion of the study without relation to the study objectives. All participants will receive current optimised treatment following established clinical guidelines on management of AF, HF and hypertension.1
Trial setting and identification of participants
The trial is coordinated by Primary Care Research and Clinical Trials Unit (PC-RCTU), University of Birmingham, including coordination of the participant searches, using clinical research network. All patients are seen, investigated and managed in the Research Clinic in the Institute of Cardiovascular Sciences (RC-ICS), City Hospital, Birmingham.
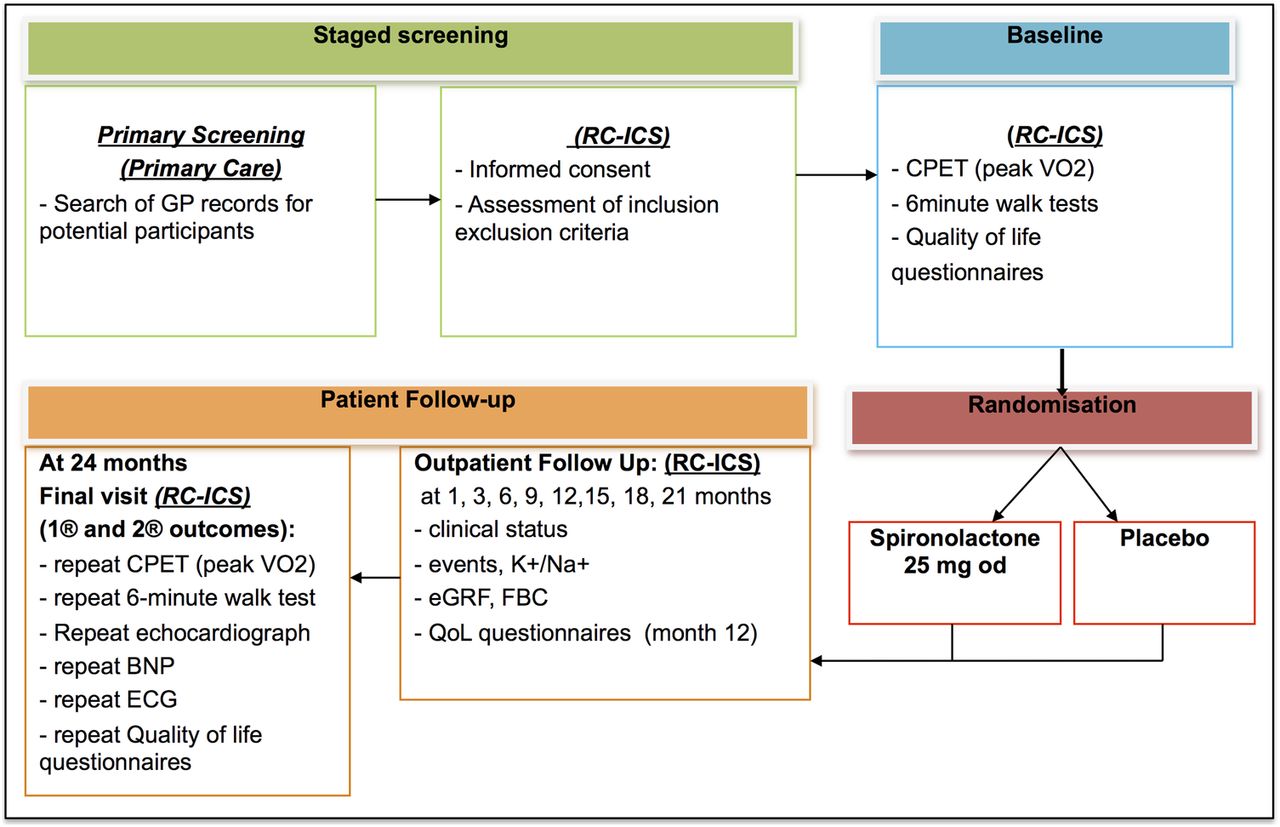
Trial participants have been recruited from primary care AF registers in family practices and outpatient AF clinics in Sandwell and West Birmingham Hospitals Trust, Birmingham. This allowed enrolment of a representative population of patients with AF. At the screening visit to the RC-ICS participants were consented into the study and screened for eligibility. During the baseline visit, the eligible patients undergone cardiopulmonary exercise testing using a cycling ergometer (to measure peak oxygen consumption (VO2)), 6 min walk test and complete quality of life questionnaires (validated Minnesota Living with Heart Failure (MLWHF)33–35 and EuroQol EQ-5D-5L (EQ-5D)36 ,37 questionnaires). After that, they were randomised into the 2-year trial. The study schema and visit schedule are shown in figure 1 and table 2.
Timeline of trial procedures alongside the assessments that will be carried out at each stage
{kind=link}
Trial schema. BNP, brain natriuretic peptide; CPET, cardio-pulmonary exercise testing; eGRF, estimated glomerular filtration rate; FBC, full blood count and haematocrit; GP, general practitioner; QoL, quality of life; RC-CCS, Research clinic of the University of Birmingham Institute for Cardiovascular Sciences, City Hospital, Birmingham, UK.
Randomisation and blinding
During randomisation (1:1), the participants were first stratified by their baseline peak VO2 (two stratification groups; VO2≤16 mL/min/kg, and VO2>16 mL/min/kg). A secure web-based randomisation system was used for the concealed allocation of a unique investigational medicinal product number to each participant. Trial participants, the trial team in contact with the patient, care providers, outcome assessors and data analysts all remain blinded to the treatment.
Blinding of the trial drug identity took place at the time of packaging and labelling (Catalent Pharma Solutions, UK). Only the database programmer and the Catalent Pharma Solutions can see the investigational medicinal product number list. A sealed copy of the list is kept to the Pharmacy Department at City Hospital (who are independent of the trial, and operate 24 hours a day). In the event of a codebreak situation occurring, the patient will be withdrawn from the trial treatment, as they will become unblinded to their trial drug.
Treatment and dosing schedule
Trial participants receive either spironolactone 25 mg once daily or matched placebo. This dose has been shown to improve outcomes in systolic HF, improve diastolic function in HFpEF and to reduce collagen turnover, a marker for fibrotic signalling, in the The Randomized Aldactone Evaluation Study (RALES) population.38 The same dose of the spironolactone within 1 year significantly improved diastolic function in participants with HFpEF from the ALDO-DHF trial.23
In the case of an increase in potassium level to 5.1–5.5 mmol/L or in the presence of other non-life-threatening side effects (such as gynaecomastia) the trial drug is downtitrated to 25 mg each second day. In such cases, the investigators are advised to reuptitrate the trial medication if the reason for downtitration has resolved.
Drug toxicity will be defined as an increase in potassium level to >5.5 mmol/L. In the case of toxicity or suspected toxicity, the trial medication will be stopped for the duration of the trial, but the patient will be requested to attend the remaining follow-up visits. Blood pressure will be controlled during the duration of the study with particular attention to blood pressure levels after beginning of the study drug and after any changes in antihypertensive agents and their doses.
Study end points
The primary efficacy end point will be the improvement in exercise tolerance at 2 years. This will be assessed by the difference between trial groups in peak VO2 on cardiopulmonary exercise testing.
The secondary efficacy end point will be the level of improvement in quality of life and diastolic function, and also the improvement the rate of all-cause hospital admissions and spontaneous return the sinus rhythm, with spironolactone. This will be assessed by: (1) improvement in exercise tolerance measured by 6 min walking test (a simple test of exercise performance) at baseline and at 2 years; (2) improvement in quality of life (MLWHF and EQ-5D36 ,37 questionnaires) over the 2-year duration; (3) improvement in left ventricular diastolic function (E/e’ ratio39–45 on echocardiography) will be assessed at baseline and at 2 years; (4) improvement in rates of all-cause hospitalisations during 2-year follow-up;35 ,36 (5) spontaneous return to sinus rhythm on ECG after 2 years of treatment. Additionally we will record any cases of major adverse clinical events, such as death from any causes, death from cardiac causes, hospitalisation for cardiac causes, a change in the New York Heart Association (NYHA) class, stroke or systemic thromboembolism.
The study started on the October 2014 and recruitment completed on 29 June 2016. We plan to complete the study by September 2018.
Statistical considerations
The analysis will follow intention-to-treat principles. The linear mixed-model analysis will be used to compare peak VO2 at 2 years between the intervention and the control group. Covariates will be peak baseline VO2, age, gender, systolic/diastolic blood pressure and body mass index measured at baseline. General practitioner practices or recruitment centres will be included as random effects.
Secondary analyses will also use linear or non-linear mixed modelling as above but with the dependent variable the secondary end points mentioned in the earlier Trial end points section. Interactions between intervention/control, age and gender will also be included in the mixed modelling analyses to see whether differences in secondary end points between intervention and control participants vary with these two factors. Missing values will be substituted using a multiple imputation procedure. Because of the likelihood of non-normality, the method of Hussain et al46 will be used.
For the primary outcome, we based our power calculation for peak VO2 on the published values of peak VO2 in participants with HF (16±5 mL/min/kg).47 We anticipate a difference of 2 mL/min/kg in the improvement in peak VO2 after 2-year treatment with spironolactone compared with the control group. Published data in HFpEF suggest that such a difference would be clinically relevant and it was factored for the design of the recent ALDO-DHF study of spironolactone in patients with HFpEF, 95% of whom were free from AF.22 ,23 ,48 Unfortunately, the study by Cicoira et al47 used for power calculation does not give a SD of the change in peak VO2 from the baseline but a similar trial, Edelmann et al49 provides that statistic (5 mL/min/kg) and also reports a similar magnitude of the effect. We estimate that a sample size of 100 participants in each arm would give the power of at least 80% to detect differences in primary and secondary end points of a magnitude consistent with published results from similar studies. The inclusion of a provision for a 20% drop out rate could potentially lead to powers of near 90% or more if the assumption of a drop rate of 20% were too pessimistic.
Study funding and management
The IMPRESS-AF trial is funded by the National Institute for Health Research (NIHR), UK. The University of Birmingham is the sponsor of this trial. The day-to-day management of the trial will be coordinated by the Primary Care Research and Clinical Trials Unit (PC-CRTU) at the University of Birmingham, registered by the NIHR as a trials unit. The Trial Management Group will meet at least monthly to ensure implementation of the trial. A Trial Steering Committee has been appointed and will be responsible for overseeing the progress of the trial. An independent Data Monitoring and Ethics Committee will be responsible for the regular monitoring of trial data and it will give advice on whether the accumulated data from the trial, together with the results from other relevant research, justify the continuing recruitment of further participants. The Data Monitoring and Ethics Committee will make confidential recommendations to the Trial Steering Committee as the decision-making committee for the trial.
Ethics and dissemination of findings
The results of the trial will be published in an international peer-reviewed journal. We hope that the study findings will inform future guidelines for management of HF.
Registration: The study is registered with European Union Clinical Trials Register (EudraCT number 2014-003702-33), clinicaltrial.gov (NCT02673463) and has been adopted by the NIHR Clinical Research Network.
Discussion
AF has a prominent role in prognostication in HF. In a recent large study of 23 644 participants with HF, of which 48.3% had documented AF, the presence of the arrhythmia was associated with higher adjusted rates of ischaemic stroke, hospitalisation for HF, all-cause hospitalisation and death irrespectively whether LVEF was impaired or preserved.50 Clinical trials of aldosterone antagonists (RALES, The Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS), The Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EMPHASIS-HF)) uniformly showed their clinical benefits in systolic HF. However, there is no established treatment for patients with AF with HFpEF.
Activation of aldosterone pathway can contribute to the progression of patients with AF to symptomatic HF despite preserved cardiac contractility, due to the promotion of cardiac fibrosis. Published evidence from AF populations supports the central role of atrial fibrosis in electrical and structural atrial remodelling, and its independent predictive value for the high risk of cerebrovascular events.51 ,52 There is an association between AF and abnormal left ventricular fibrosis, which related to the depressed diastolic function in such participants.53
According to a substudy of the RALES trial, the improved survival in participants treated by spironolactone was linked to its ability to reduce serum markers of ongoing fibrosis (type I and III collagen synthesis).38 Additionally, aldosterone leads to cardiac invasion by proinflammatory mononuclear cells.54 Aldosterone antagonists (ie, spironolactone or eplerenone) ameliorate left ventricular fibrosis in animal models and reduce levels of serum markers of collagen turnover in humans with HFpEF (n=44).55 ,56 In a small, published pilot trial, spironolactone reduced left ventricular fibrosis and improved diastolic function in participants with HFpEF (dilated cardiomyopathy, n=25).57
The randomised IMPRESS-AF study should help understanding utility of aldosterone inhibition in permanent AF for prevention of deterioration or improvement in exercise tolerance and quality of life as well as in cardiac diastolic function.
Acknowledgments
The authors would like to acknowledge and thank Dr Farhan Shahid, Dr Christos Voukalis, Sister Rebecca Brown and Mr Andrew Cooley for patient recruitment and care in the University of Birmingham Institute for Cardiovascular Sciences, City Hospital; the whole team of the Primary Care Research and Clinical Trials Unit, University of Birmingham for the trial management; the Clinical Research Network West Midlands for identification and approaching prospective participants. The authors would like to express a special gratitude to the members of the Trial Steering Committee: Professor Diana Adrienne Gorog, Dr Andrew Appelboam, Dr Sajjad Sarwar, Renton Caroline, and members of Data Monitoring and Ethics Committee: Dr Derick Todd, Dr Paul Ewings and Norman Paul Briffa.
References
Footnotes
Contributors ES and GYHL provided the study hypothesis, and protocol development. ES wrote the first draft, with assistance from GYHL. Other authors developed and refined the study protocol. All authors provided critical revision of study protocol.
Funding This work was supported by NIHR-EME Program, Researcher led grant 12/10/19.
Competing interests None declared.
Ethics approval National Research and Ethics Committee West Midlands—Coventry and Warwickshire (REC Reference 14/WM/1211).
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement This is a trial design paper from a trial in progress. The trial results will be available after completion of the trial.