Article Text

Abstract
Objectives Facioscapulohumeral muscular dystrophy type 1 (FSHD1) has been genetically linked to reduced numbers (≤8) of D4Z4 repeats at 4q35. Particularly severe FSHD cases, characterised by an infantile onset and presence of additional extra-muscular features, have been associated with the shortest D4Z4 reduced alleles with 1–3 repeats (1–3 DRA). We searched for signs of perinatal onset and evaluated disease outcome through the systematic collection of clinical and anamnestic records of de novo and familial index cases and their relatives, carrying 1–3 DRA.
Setting Italy.
Participants 66 index cases and 33 relatives carrying 1–3 DRA.
Outcomes The clinical examination was performed using the standardised FSHD evaluation form with validated inter-rater reliability. To investigate the earliest signs of disease, we designed the Infantile Anamnestic Questionnaire (IAQ). Comparison of age at onset was performed using the non-parametric Wilcoxon rank-sum or Kruskal-Wallis test. Comparison of the FSHD score was performed using a general linear model and Wald test. Kaplan-Meier survival analysis was used to estimate the age-specific cumulative motor impairment risk.
Results No patients had perinatal onset. Among index cases, 36 (54.5%) showed the first signs by 10 years of age. The large majority of patients with early disease onset (26 out of 36, 72.2%) were de novo; whereas the majority of patients with disease onset after 10 years of age were familial (16, 53.3%). Comparison of the disease severity outcome between index cases with age at onset before and over 10 years of age, failed to detect statistical significance (Wald test p value=0.064). Of 61 index cases, only 17 (27.9%) presented extra-muscular conditions. Relatives carrying 1–3 DRA showed a large clinical variability ranging from healthy subjects, to patients with severe motor impairment.
Conclusions The size of the D4Z4 allele is not always predictive of severe clinical outcome. The high degree of clinical variability suggests that additional factors contribute to the phenotype complexity.
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Strengths and limitations of this study
This is the most comprehensive survey of the clinical status of patients carrying a 1–3 D4Z4 reduced allele (DRA). Data were acquired through the Italian National Registry for facioscapulohumeral muscular dystrophy (FSHD) (INRF), which systematically collects clinical and molecular data from the whole Italian territory.
The Infantile Anamnestic Questionnaire (IAQ) form was designed to acquire retrospective information about pregnancy, delivery, birth and perinatal period of carriers of 1–3 DRA.
Mixed methods were used to obtain a standardised clinical assessment of all participants, including questionnaire, interview data and a structured medical assessment undertaken by the Italian Clinical Network for FSHD (ICNF).
Data were usually self-reported for the medical history, without access being sought to individuals’ health records.
The data sets were derived only from Italy and have their own limitations.
Introduction
Facioscapulohumeral muscular dystrophy (FSHD (MIM 158900)) is the third most common muscular dystrophy with an estimated prevalence of 1:20 000.1 FSHD is considered an autosomal dominant disorder, with a typical onset within the second decade of life.2 ,3 The disease presents a remarkably wide variety of phenotypic expressions, ranging from almost asymptomatic subjects to severe wheelchair-dependent patients.4–6 The classical FSHD phenotype, first described as an independent nosological entity in 1884, by Landouzy and Dejerine,7 is characterised by progressive facial, shoulder girdle and pectoral muscle weakness and atrophy. Disease progression may lead to involvement of abdominal and pelvic muscles, causing lumbar hyperlordosis and a waddling gait. Weakness of anterior leg muscles results in a steppage gait.
Several genotype–phenotype correlation studies reported a rough inverse correlation between the number of D4Z4 repeats and the severity of FSHD.5 ,8–10 It has thus been suggested that alleles of extremely short size (1–3 D4Z4 repeats) are associated with the most severe form of disease.5 ,6 ,8 ,11 A number of reports described cases carrying very short D4Z4 alleles with 1–3 D4Z4 repeats characterised by childhood onset, rapid progression of muscle weakness and extra-muscular clinical features.12–20 In 1994, Brouwer et al21 introduced the concept of Infantile Onset FSHD, based on the following diagnostic criteria: (1) signs or symptoms of facial weakness by 5 years of age; (2) signs or symptoms of scapular girdle weakness by 10 years of age.
However, several exceptions to these general trends have been found since the molecular analysis of the D4Z4 region became part of clinical diagnoses. Differences of clinical expression have been documented between participants carrying shorter alleles, varying from very severe forms of disease and complex phenotypes starting in infancy,12–20 to milder form or asymptomatic carriers.6 ,22–25 Based on the results presented in these studies, it was not possible to establish whether a congenital form of FSHD exists and whether detection of a 1–3 D4Z4 reduced allele (DRA) is always predictive of a severe phenotype with infantile onset. Furthermore, it is unclear whether additional clinical features observed in some patients with FSHD represent the extreme of the FSHD clinical spectrum or if they result from random associations.
In the present study, we investigated the prognostic significance of very short 4q35 alleles (1–3 DRA), through the clinical evaluation of 66 index cases and 33 relatives. Moreover, we searched for signs of perinatal onset through the systematic collection of anamnestic records of 80 patients carrying 1–3 DRA. Our study aimed to examine the clinical variability in the cohort of the participants carrying the shortest 4q35 alleles, presented in earlier clinical studies,6 ,26–28 supporting the hypothesis that additional factors must contribute to FSHD disease.
Methods
Study design and subject selection
The study was performed on FSHD families accrued through the Italian Clinical Network for FSHD (ICNF) (http://www.fshd.it).29 The INCF is distributed across all of Italy, and includes a diagnostic laboratory at the University of Modena and Reggio Emilia, and 14 clinical centres, networking with the Italian Association of Myology (http://www.miologia.org). All clinical and molecular data were collected in the Italian National Registry for FSHD (INRF) database at the Miogen Laboratory at the University of Modena. The present study was conducted from January 2008 to December 2013. Of 850 index cases from the INRF, we identified 114 index cases carrying DRA with 1–3 repeats (figure 1A) and fulfilling the clinical diagnostic criteria defined for FSHD.30 Family studies were conducted in 66 index cases, in which clinical and molecular analysis was extended to all available relatives willing to participate. Screening for 1–3 DRA was performed in 226 relatives. We defined de novo cases as single participant with neither parent carrying DRA; when the DRA was detected in one of the parents and/or other family members (ie, sibs), we classified the participant as familial. We considered participants as not informative when it was not possible to examine their parents and/or other informative family members.
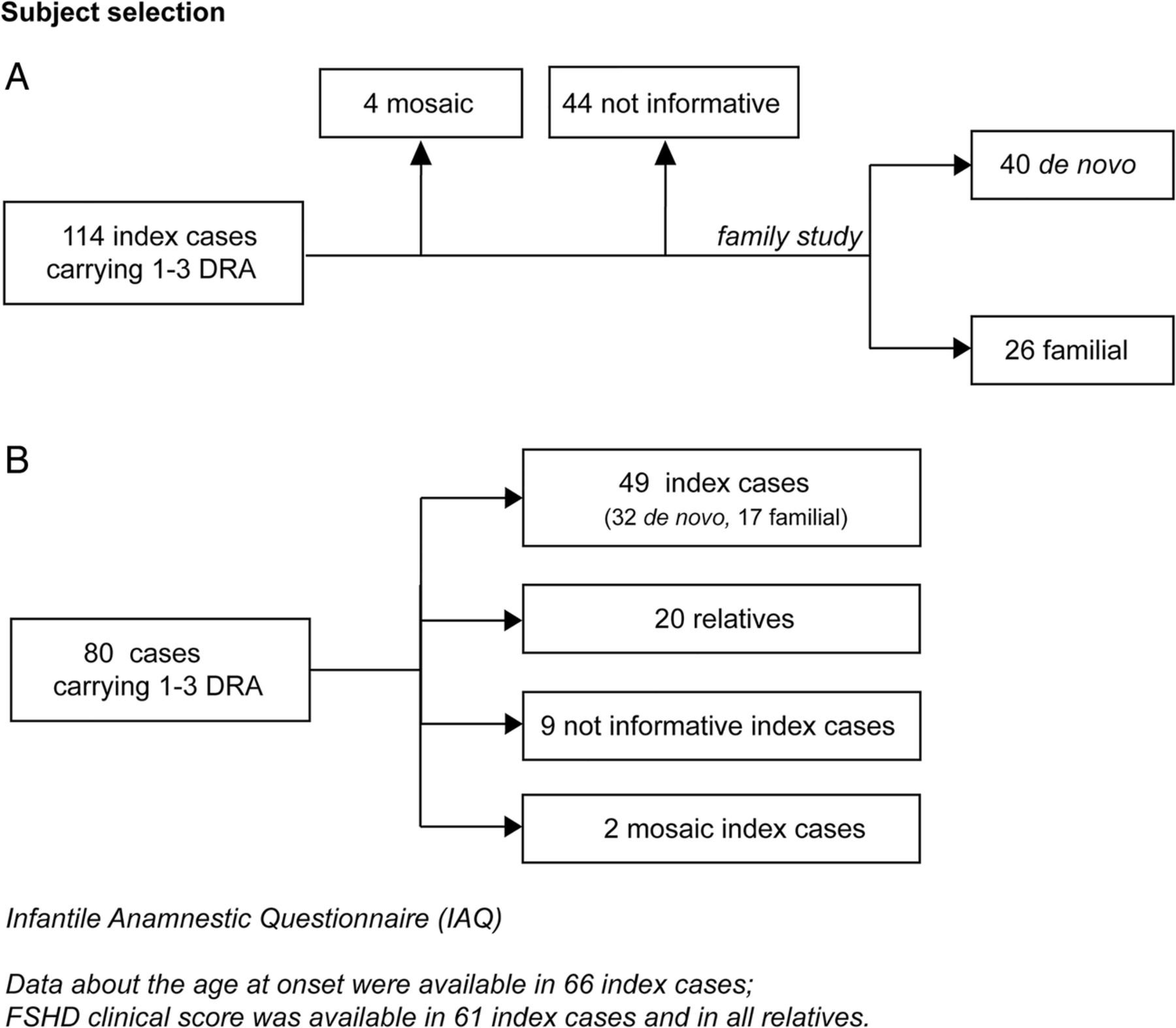
Selection of probands from the Italian National Registry of facioscapulohumeral muscular dystrophy (FSHD) for clinical and molecular study. (A) Data on age at onset were available for all index cases carrying 1–3 D4Z4 reduced alleles (DRA); FSHD clinical score was assessed in 61 index cases and 33 relatives carrying 1–3 DRA, recruited for the family study. (B) The Infantile Anamnestic Questionnaire was administered in 80 cases carrying 1–3 DRA.
Informed consent, according to the Declaration of Helsinki, was obtained from each participant enrolled in the study.
Clinical examination
The clinical examination was performed using the standardised FSHD clinical protocol with validated inter-rater reliability.29 The FSHD clinical protocol was developed by the ICNF in order to numerically define the clinical severity of the motor impairment. The FSHD score, which translates disability into a number, ranges from zero, when no objective evidence of muscle functional impairment is present, to 15, when all the muscle groups tested are severely impaired (http://www.fshd.it). Age at onset was estimated on the basis of patients’ records. To obtain a more objective evaluation of facial weakness onset, we asked specific questions, such as, ‘Have your relatives ever noticed that you were sleeping with half-closed eyes?’; ‘Have you ever been able to drink with a straw?’; ‘Have you ever noticed difficulty in blowing out candles?’ (see online supplementary figure S1). When participants did not report of motor impairment, but a mild muscle weakness was observed during clinical examination, we arbitrarily considered the age at examination as the age at onset, according to previous reports.6 ,31
Data about age at onset were available for all index cases, while the clinical re-evaluation using FSHD clinical score was performed in the 61 index cases and 33 relatives recruited for the family study (figure 1A,B).
In order to investigate the earliest signs of disease and to rule out pre- or perinatal events as possible causes of delayed achieving of motor milestones, we designed the Infantile Anamnestic Questionnaire (IAQ) (see online supplementary figure S1). All data about: (1) pregnancy, (2) birth, (3) the prenatal period and first month of life and (4) psychomotor and language development were collected in a retrospective manner. Items related to each section were scored as normal/altered. We collected anamnestic reports about neurological examinations in the first year of life, together with clinical and instrumental data in the following years, whenever possible, in 80 cases carrying 1–3 DRA (figure 1B).
Molecular characterisation
DNA was prepared from isolated lymphocytes according to standard procedures. In brief, restriction endonuclease digestion of DNA was performed in agarose plugs with the appropriate restriction enzyme: EcoRI, EcoRI/BlnI. Digested DNA was separated by pulsed field gel electrophoresis (PFGE) in 1% agarose gels, as previously described.28
Allele sizes were estimated by Southern hybridisation with probe p13E-11 of 7 μg of EcoRI-digested, EcoRI/BlnI-digested genomic DNA extracted from peripheral blood lymphocytes, electrophoresed in a 0.4% agarose gel, for 45–48 h at 35 V, alongside an 8–48 kb marker (Bio-Rad) (see online supplementary figure S2A). Participants carrying alleles of 11–19 kb (1–3 D4Z4 units) in size were included in the study (see online supplementary figure S2B). To distinguish whether the DRA came from chromosome 10q or from 4q, DNA from each proband was analysed by NotI digestion and hybridisation with the B31 probe, to confirm the chromosome 4q origin of the 11–19 kb EcoRI allele.28 Restriction fragments were detected by autoradiography or by using a Typhoon Trio system (GE Healthcare).
Statistical analysis
Comparison of age at onset was performed in de novo, familial and relatives of index cases using the non-parametric Wilcoxon rank-sum (for comparison between two groups) or Kruskal-Wallis (for comparison among three groups) test. To calculate the prevalence of infantile onset, we subdivided the patients on the basis of the age at onset, before 10 years and after 10 years of age. The prevalence of infantile onset was then compared across de novo, familial and relatives of index cases using χ2 test.
Comparison of the FSHD score among de novo, familial and relatives of index cases was performed using a general linear model adjusted by sex and age at examination. Wald tests were used to evaluate if FSHD score differs between de novo, familial and relatives of index cases.
For the cohorts of probands and family members, time before developing disease was estimated from birth to the earliest age at onset of motor impairment. Kaplan-Meier survival analysis was used to estimate the age-specific cumulative motor impairment risk and loss of independent walking,32 with the corresponding 95% CI. The differences in cumulative risk between de novo and familial index cases, carriers of 1–3 DRA, were evaluated using the Log rank test.
Results
High percentage of de novo rearrangements among carriers of 1–3 DRA
Molecular analysis of 114 index cases from the INRF revealed that four participants were carriers of a somatic mosaicism for DRA. Forty-four cases were considered as not informative, since parents and/or other family members were not available for the molecular investigation. In 66 unrelated index cases, we extended molecular characterisation to parents and/or other relatives and found that 26 probands were familial (39.4%) and 40 probands were de novo (60.6%). We also established the parental origin of de novo rearrangements in 31 cases and found that 48.4% were from the father and 52.6% were from the mother. Parental age at conception ranged from 18 to 43 years (maternal mean age at conception: 28.1±5.1 years; paternal mean age at conception: 31.3±5.3). In neither de novo cases nor in familial cases did we observe any correlation between the detection of a de novo D4Z4 rearrangement at 4q with the parent's age at conception.
Our analysis shows that the percentage of de novo cases in our cohort is higher (60.6%) than that previously described in the whole FSHD1 population (10–30%).9 ,10 ,33–37 However, when we considered the 246 probands with 4–8 DRA we found that only 14 cases carried a de novo DRA (5.7%). Therefore, the overall percentage of de novo cases (17.3%) observed in the Italian cohort of 312 FSHD1 probands does not differ from published records. Instead, our analysis shows a skewed percentage of carriers of de novo rearrangement in the cohort of 1–3 DRA carriers, rather than in the cohort of 4–8 DRA carriers.
FSHD onset before 10 years of age is more prevalent among de novo carriers
Several studies suggest that carriers of 1–3 DRA develop a severe form of disease. In particular, the age at onset has been considered the clinical feature discriminating different FSHD phenotypic entities,21 and has been used as a prognostic parameter for defining phenotype severity.31 In a previous study, we found that FSHD occurs earlier in families in which 1–3 DRA segregate.6 In the present study, we extended our analysis to all probands carrying 1–3 DRA, subdivided in two groups, de novo and familial. Table 1A shows that the mean age at onset observed among de novo probands appears significantly earlier than in familial.
Age at onset among de novo and familial index cases carrying 1–3 DRA
To verify the prevalence of infantile onset, we then subdivided these two groups of patients on the basis of the age at onset, before 10 years and after 10 years of age (table 1B). In 54.5% of index cases carrying 1–3 DRA, the disease onset was in the first decade of life. A higher percentage of these cases with infantile onset reported facial weakness as the most common sign of disease (76.5%), while shoulder girdle weakness at onset resulted most commonly (69%) among patients developing FSHD after 10 years of age. Notably, the majority of patients with early disease onset were de novo. In contrast, the majority of patients with disease onset after 10 years of age were familial. We then estimated the age-specific cumulative motor impairment incidence (see online supplementary figure S3A), with the corresponding 95% CI in the two cohorts of de novo and familial probands by Kaplan-Meier survival analysis. For each individual, we assessed the interval of time from birth to the earliest age at onset of motor impairment. Supplementary figure S3A shows that, among participants carrying a de novo 1–3 DRA, the risk of developing motor impairment is 65% by age 10 years, 88% by age 15 years and 98% by age 20 years. Among participants carrying a familial 1–3 DRA the risk is 38% by age 10 years, 77% by age 15 years and 88% by age 20 years. Therefore the risk of developing FSHD before 10 years of age is significantly higher in participants carrying a de novo 1–3 DRA.
In our previous work6 on the large cohort of FSHD1 families, we showed that the risk of developing motor impairment is higher in male probands/relatives during adulthood (18–55 years), and is similar between males and females in childhood-teens and in the elderly. In this cohort of patients carrying 1–3 DRA, the cumulative risk of motor impairment according to sex did not show significant difference among males and females (log rank test p value=0.305). We hypothesise that the absence of gender difference is due to the fact that, in this group, the disease onset occurs, on average, before patients reach 20 years of age.
Infantile onset does not always predict a very severe clinical outcome
It has been reported that extremely short 4q35 alleles (1–3 D4Z4 units) are associated with more severe forms of FSHD.5 ,6 ,8 ,11 Considering the earlier average onset in de novo cases compared to familial 1–3 DRA carriers, we tested whether de novo index cases present a more severe disease expression, comparing the degrees of motor disability indicated by the FSHD score.29 Statistical evaluation failed to detect any significant difference in the mean FSHD score adjusted by age and sex between the two groups (10.6 in de novo index cases vs 9.5 in familial index cases; 95% CI, respectively, (8.0 to 11.3), (9.3 to 12.0); Wald test p value=0.280).
We then calculated the age-specific relative risk of loss of independent walking ability in de novo familial index cases, using Kaplan-Meier statistics. This analysis did not detect a statistically significant difference between the two groups (see online supplementary figure S3B), although the percentage of wheelchair-bound patients under 40 years of age is higher in de novo index cases (70%) versus familial index cases (35%). Finally, comparison of the severity of disease expression between index cases (both de novo and familial) with early age at onset (0–10 years; age and sex adjusted mean FSHD score=11.4) versus probands with age at onset over 10 years (age and sex adjusted mean FSHD score=9.5) failed to detect statistically significant more severe FSHD disease in probands with infantile onset (Wald test p value=0.064). Overall, the lack of significant differences in each of these three comparisons argues against the idea that early FSHD onset is necessarily associated with a more disabling outcome.
Relatives carrying 1–3 DRA are not always severely affected
In eight families, we were able to extend molecular analysis to three generations, and found that the molecular defect appeared de novo and was transmitted to offspring in six members. Three carriers of de novo mutation were probands, whereas three other carriers were discovered because of the appearance of FSHD in one child. Importantly, two of them were unaffected (see online supplementary figure S4) at the time of examination (41 and 45 years, respectively) and one suffered from a mild form of disease (FSHD score 3 at 38 years of age). These observations indicate that the presence of a de novo 1–3 DRA does not always associate with a severe phenotype.
We compared the age at disease onset detected in the group of familial probands with that recorded in the group of 33 relatives carrying a DRA. This comparison displayed that affected relatives had significantly later onset of FSHD than the probands (table 2A).
Age at onset in familial index cases and their relatives carrying 1–3 DRA
We also compared the degree of motor impairment, recorded as FSHD score, detected in the two groups. The age and sex adjusted mean FSHD score received by relatives was significantly lower than that recorded in the probands (4.7 vs 9.5; Wald test p value <0.0001). Notably, four relatives (12.1%), respectively aged 33, 42, 47 and 50 years, presented no muscle weakness (table 2B). Overall, similarly to what we observed among carriers of 4–8 DRA,6 we found a reduced severity of clinical expression also in the group of relatives carrying very short D4Z4 allele in comparison with probands, including participants with no signs of disease after the age of 40 years.
Carriers of 1–3 DRA did not show signs of prenatal and neonatal FSHD onset
To systematically obtain information about the perinatal period and the appearance of the first signs and/or symptoms of disease, we designed the IAQ (see online supplementary figure S1). We gathered anamnestic data about pregnancy, delivery and birth from all participants who were able to respond to this questionnaire. We interviewed 80 cases carrying 1–3 DRA (figure 2). Figure 2 shows that no significant alterations in pregnancy, delivery and birth were reported. There was no report of any floppy infant at birth. In 72 of 80 participants (90%), psychomotor development milestones were reached appropriately.

Infantile anamnestic records of 80 carriers of 1–3 D4Z4 reduced alleles (DRA) (NA, not applicable).
This analysis shows that children carrying 1–3 DRA do not display signs of muscle weakness prenatally or at birth. Moreover, signs that can possibly be attributed to early onset of muscle weakness are reported only in a small percentage of participants. Therefore we conclude that very early onset is not a frequent feature of FSHD.
Carriers of 1–3 DRA with extra-muscular clinical conditions
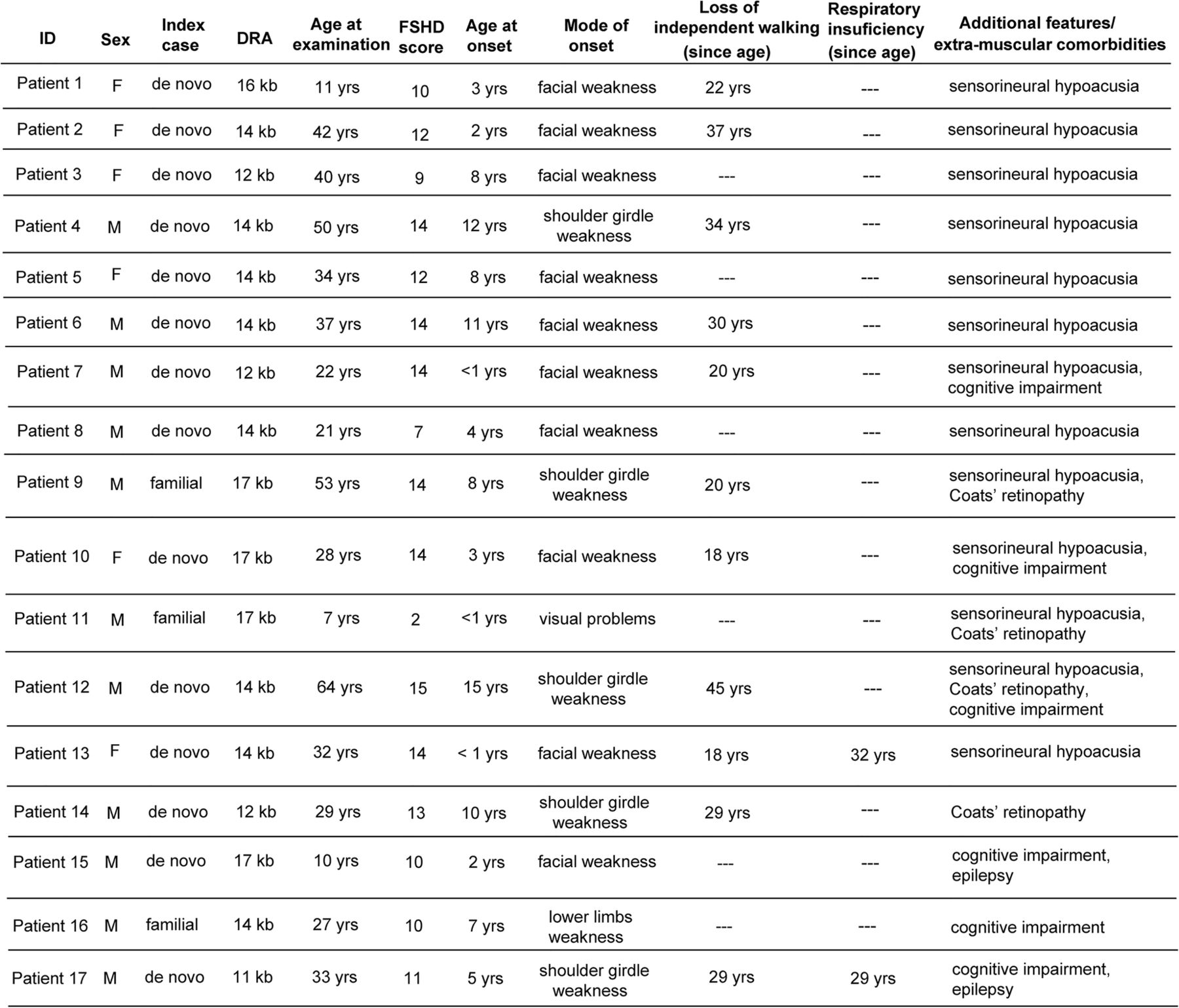
In some reports, severe FSHD is associated with extra-muscular features such as sensorineural deafness, Coats’ retinopathy, epilepsy and mental retardation. We assessed the frequency of additional clinical conditions in the cohort of 61 index cases carrying 1–3 DRA, summarised in figure 3.

Clinical features of 1–3 D4Z4 reduced alleles (DRA) carriers with extra-muscular comorbidities (FSHD, facioscapulohumeral muscular dystrophy; M, male; F, female).
Thirteen participants suffered from sensorineural deafness (21.3%). In eight cases, it was isolated, with no other recognisable medical condition, and in five cases we detected additional extra-muscular manifestations.
In four cases, we observed Coats’ retinopathy (6.6%). In one it was found as an isolated condition, whereas in three other cases it was associated with sensorineural deafness or cognitive impairment. Cognitive impairment was reported in six cases (9.8%), and two of these also suffered from epilepsy. All cases with mental retardation showed a very severe form of disease.
We also investigated the presence of restrictive respiratory disease requiring intervention, previously described in about 1% of patients with FSHD1, typically in patients with severe muscle weakness.3 In our cohort of 61 index cases with 1–3 DRA, we identified 7 cases (11.5%) with respiratory insufficiency requiring non-invasive ventilation (NIV). Two of these showed a complex phenotype with additional extra-muscular manifestations (figure 3), while all the others suffered from a disabling form of disease and required use of NIV since the ages of 22, 29, 30, 42 and 53 years, respectively.
Discussion
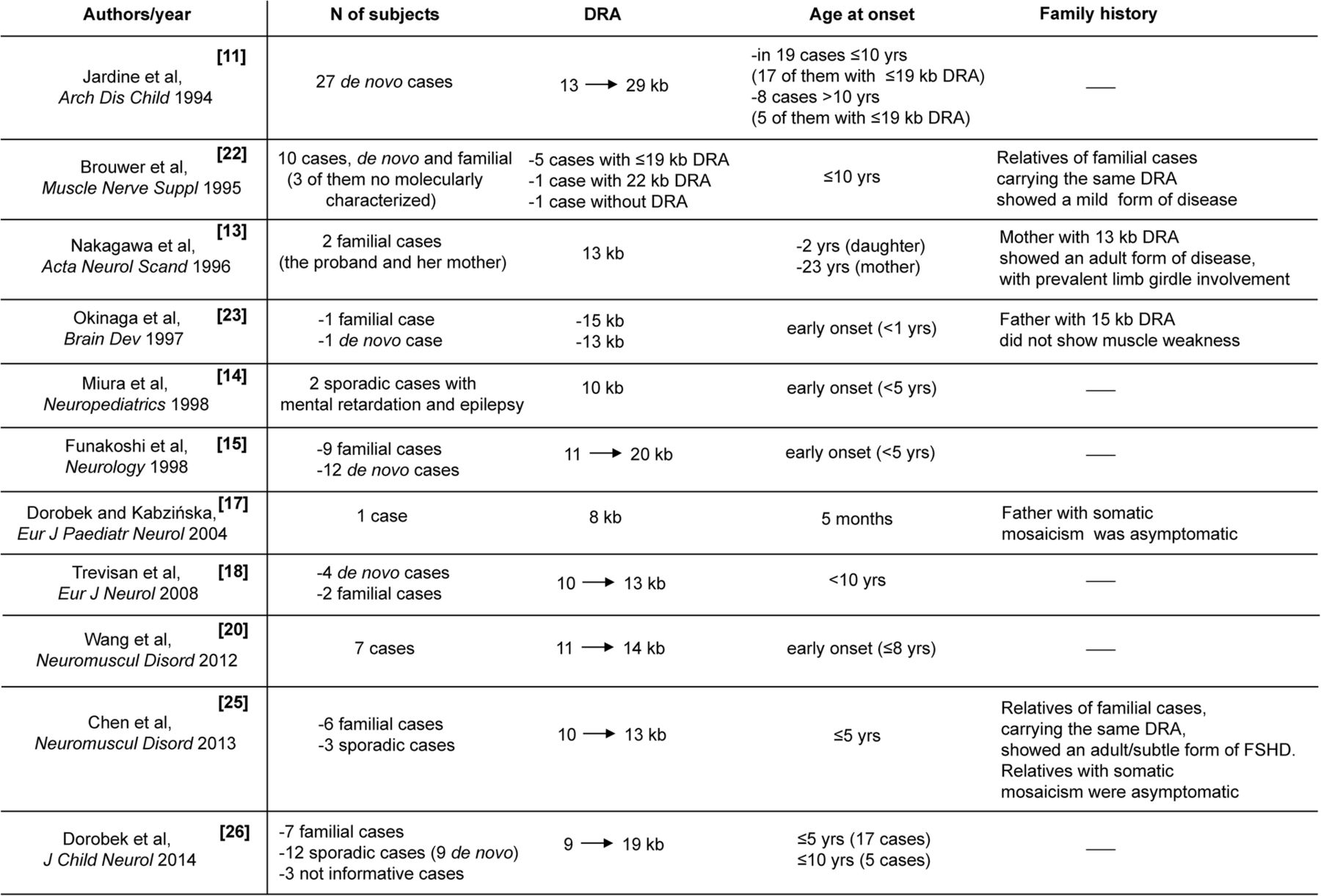
Since molecular analysis of the D4Z4 region was introduced to study FSHD, it has been suggested that very severe forms of disease are associated with a very short DRA.5 ,6 ,8 ,11 This notion has supported the idea that a rough inverse correlation exists between the size of D4Z4 allele and disease severity. Moreover, it has been long debated whether ‘infantile FSHD’ might exist as a distinct nosological entity, characterised by specific peculiarities that distinguish it from classical FSHD with onset in the second decade of life.21 ,22 ‘Infantile FSHD’ has been defined by childhood onset and severe muscle impairment, associated with high-frequency hearing loss, retinal vascular abnormalities, mental retardation and epilepsy.14 ,15 ,18 ,20 By revising the literature, as summarised in figure 4, we found that not all severe cases had an infantile onset, and not all carried a very reduced size D4Z4 allele. However, the different designs used in each of these previous studies prevented the possibility of pooling various observations to obtain a complete or more defined picture of clinical features of participants carrying ‘very short’ D4Z4 allele.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Revisited literature: 1–3 D4Z4 reduced alleles (DRA) case reports comorbidities (FSHD, facioscapulohumeral muscular dystrophy).
Here, the large number of index cases carrying DRA with 1–3 units accrued through the INRF allowed us to obtain more precise information about this group of patients. In particular, we aimed at verifying whether perinatal onset of FSHD exists and whether presence of a 1–3 DRA is always predictive of a severe phenotype associated with infantile onset. In addition, we searched this cohort of patients for the presence of additional clinical conditions determining complex phenotypes.
First, our analyses showed that the majority of 1–3 DRA carriers (60.6%) are de novo. These data, together with the observation that 1–3 DRAs have never been detected in the normal population,28 support the notion that the D4Z4 repeat array is highly recombinogenic and therefore prone to a high mutation rate.35 ,37
Second, the majority (72.2%) of cases presenting disease onset before 10 years of age are isolated and carry a de novo rearranged DRA; in contrast, the majority (53.3%) of familial FSHD1 cases develop around the second decade of life (table 1B). However, we did not find a statistically significant difference in disease outcome between de novo and familial probands, even though there is a trend towards a more severe progression among de novo cases (age and sex adjusted mean FSHD score in de novo vs familial probands, is 10.6 vs 9.5). We confirm a more severe phenotype in index cases carrying 1–3 DRA in comparison with participants carrying longer alleles (4–10 DRA).6 ,38 Our data also confirmed that, among index cases with infantile onset, the majority (76.5%) reported facial weakness as the first sign of disease, presenting difficulty in closing eyes and puffing cheeks, or progressive facial hypomimia. In three participants, we collected anamnestic records of abnormalities in pronouncing some phonemes at the age of 3–4 years, and interpreted these difficulties as possibly due to the onset of facial muscle weakness. Two cases were initially misdiagnosed as affected by Moebius syndrome, most likely because of the very early onset of severe facial muscle weakness. Instead index cases with FSHD onset after 10 years reported shoulder girdle weakness as the most common first sign of the disease, according to the previous reports.6 ,39 ,40
Third, even when signs or symptoms of muscle weakness are detected within the first year of life, the long-term disease outcome does not differ from cases with later onset.
Fourth, the use of the IAQ confirmed that pre- and perinatal onset is not present in the group of 1–3 DRA carriers.
Fifth, among the 33 relatives carrying 1–3 D4Z4 alleles, 4 (12.1%) were unaffected, confirming the incomplete penetrance also in the cohort of participants carrying shorter DRA.6 Moreover, relatives displayed a milder phenotype than family proband, supporting the notion that in this subgroup of patients, the genetic background also plays a role in modulating the disease expression.6
Finally, in contrast with the previous reports,11–19 our investigations demonstrated that only seven cases (11.5%; 5 de novo and 2 familial) displayed extra-muscular comorbidities (Coats’ retinopathy, sensorineural deafness, mental retardation, epilepsy) in various combinations (figure 4). We thus conclude that extra-muscular clinical features are not part of a specific nosological entity associated with 1–3 DRA.
In summary, our study shows that a high variability of FSHD clinical expression is also present among participants carrying 1–3 DRA, with some healthy relatives carrying the same DRA as the affected ones. The observation that the majority of 1–3 DRA cases carry ‘de novo’ rearrangements, confirms the high frequency of recombination events within the D4Z4 region. Importantly, our comparisons of probands and relatives disclose that the presence of a de novo 1–3 DRA is not always associated with a disease phenotype, and emphasises the possibility that a more disabling phenotype might have a negative influence on reproductive fitness. Accordingly, in our cohort de novo rearrangements in unaffected individuals or in patients displaying mild phenotypes had normal reproductive fitness with transmission of the DRA to the offspring.
Our analysis, conducted on the largest cohort of 1–3 DRA carriers to date, shows that 1–3 DRAs are not always predictive of infantile onset or severe disease outcome. Importantly, the finding that only 27.9% of 1–3 DRA carriers present extra-muscular clinical conditions supports the notion that additional defects contribute to a more complex clinical phenotype.
Overall, our study indicated that an important future goal of FSHD clinical research is the selection of patients with homogeneous clinical features, regardless of the size of D4Z4 alleles, to provide the appropriate background for molecular studies aimed at dissecting the complex pathogenesis of this disease.
Acknowledgments
The authors are thankful to all the patients with FSHD and their families who participated in this study. The authors thank Paul D Kaufman and Mayana Zatz for their in-depth critique of the manuscript. The authors are greatly indebted to Hanna Lachert and the Segal family, for supporting our research with their generous donations.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
Footnotes
Contributors AN contributed to study design, molecular analysis, data collection, data analysis and interpretation, literature search, preparing of figures/tables and manuscript writing. GR contributed to study design, recruitment of patients, clinical evaluation, data analysis and interpretation, literature search and manuscript writing. FS contributed to study design, statistical analysis and data interpretation. EB contributed to recruitment of patients, clinical evaluation and data collection. FB and MG contributed to molecular analysis and data collection. MR contributed to recruitment of patients, clinical evaluation and data collection. SR, GB, CF, LV, LM, MC and MCD contributed to recruitment of patients and clinical evaluation. GS, GA, LS, TM, MM, LM, CA, ADM, CR, GT, MGD, LR and CB contributed to recruitment of patients, clinical evaluation and data interpretation. AB contributed to study design, clinical evaluation, data interpretation and manuscript writing. RT contributed to study design, molecular analysis, clinical evaluation, data interpretation, literature search and manuscript writing.
Funding This work was supported by Telethon Italy GUP11009, Telethon Italy GUP13012 and by Association Francaise contre les Myopathies (AFM) grant number 14339. AN was supported by the UE Initial Training Network Project number 238242 “DISCHROM”.
Competing interests None declared.
Patient consent Obtained.
Ethics approval Local ethics committees of all participating institutions.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement No additional data are available.