Article Text

Abstract
Introduction In acute pulmonary embolism (PE), poor outcome is usually related to right ventricular (RV) failure due to the increase in RV afterload. Treatment of PE with RV failure without shock is controversial and usually relies on fluid expansion to increase RV preload. However, several studies suggest that fluid expansion may worsen acute RV failure by increasing RV dilation and ischaemia, and increase left ventricular compression by RV dilation. By reducing RV enlargement, diuretic treatment may break this vicious circle and provide early improvement in normotensive patients referred for acute PE with RV failure.
Methods and analysis The Diuretic versus placebo in Pulmonary Embolism with Right ventricular enlargement trial (DiPER) is a prospective, multicentre, randomised (1:1), double-blind, placebo controlled study assessing the superiority of furosemide as compared with placebo in normotensive patients with confirmed acute PE and RV dilation (diagnosed on echocardiography or CT of the chest) and positive brain natriuretic peptide result. The primary end point will be a combined clinical criterion derived from simplified Pulmonary Embolism Severity Index (PESI) score and evaluated at 24 h. It will include: (1) urine output >0.5 mL/kg/min for the past 24 h; (2) heart rate <110 bpm; (3) systolic blood pressure >100 mm Hg and (4) arterial oxyhaemoglobin level >90%. Thirty-day major cardiac events defined as death, cardiac arrest, mechanical ventilation, need for catecholamine and thrombolysis, will be evaluated as a secondary end point. Assuming an increase of 30% in the primary end point with furosemide and a β risk of 10%, 270 patients will be required.
Ethics and dissemination Ethical approval was received from the ethical committee of Ile de France (2014-001090-14). The findings of the trial will be disseminated through peer-reviewed journals, and national and international conference presentations.
Trial registration number NCT02268903.
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Background and rationale
Acute pulmonary embolism (PE) is a frequent and serious disease.1 Its incidence ranges between 70 and 98 cases/100 000 persons per year. Early mortality associated with PE ranges from 7% to 11%, which in Europe corresponds to at least one million deaths per year.2–4 Outcome of acute PE is mainly related to haemodynamic status and to the occurrence of acute right ventricular (RV) failure due to RV pressure overload.5 ,6 Consequently, current guidelines edited by the European Society of Cardiology (ESC) and the American Heart Association (AHA) recommend these two parameters for stratifying seriousness of PE.7 ,8 These two parameters allow 30 days prognosis assessment and, therefore, they are used to determine optimal treatment. The high-risk group (mortality exceeding 15%) is represented by patients with cardiogenic shock. Patients without shock but with acute RV dysfunction and/or evidence of RV damage constitute the intermediate risk group for the ESC (submassive PE for the AHA) with a mortality rate ranging between 3% and 15%.7 ,8 RV dysfunction is defined by dilation, hypokinesis or pressure overload on echography, RV dilation on spiral CT or brain natriuretic peptide (BNP) or NT-pro BNP increase, and injury is defined by positive cardiac troponin by both the ESC and the AHA.7 ,8 Curative treatment of PE relies on anticoagulant therapy, with unfractionated heparin, low molecular weight heparin, fondaparinux or new oral anticoagulants. In patients with high-risk PE, thrombolysis is indicated in association with unfractionated heparin,7 whereas the role of thrombolysis is much more controversial in patients with intermediate-risk PE, because of its associated risk of bleeding.9 ,10
Regarding symptomatic treatment, the challenge is to overcome RV failure. Indeed, RV plays a key role in maintaining haemodynamic stability during PE. RV dysfunction is present in 15–40% of patients during acute PE, and results from increased wall stress and cardiac ischaemia, both related to pressure overload.11 The initial abrupt increase in RV afterload decreases RV stroke volume and cardiac output.12 The compensatory maintenance of cardiac output is achieved by a combination of catecholamine-driven tachycardia and preload increase through a Frank-Starling mechanism that basically relies on the RV chamber dilation.13 ,14 However, RV radius enlargement will further increase the RV wall stress, and cause a paradoxical septal motion that may impair left ventricular (LV) compliance and decrease cardiac output.15 Despite an initial haemodynamic benefit, RV dilation becomes deleterious and a vicious cycle is established between RV enlargement and the decrease of cardiac output. Thus, normotensive patients with RV dilation or injury have poorer clinical outcome with 10% PE-related shock and 5% in-hospital death.7 The management of these patients is not clearly established. Symptomatic therapy consisting of a modest volume expansion is sometimes recommended to prevent PE-related shock. Mercat et al16 showed that a 500 mL fluid expansion in acute PE improves cardiac output, nevertheless, this improvement was mainly obtained in patients with modest RV dilation. Other studies suggest that volume expansion may worsen RV function by causing a mechanical overstretch that increases RV wall stress and a septal paradoxical motion that increases LV filling pressure and reduces cardiac output.15 ,17–19 These data suggest that in patients with severe RV dilation, fluid loading should be avoided, and therapy that aims to reduce RV preload and wall stress, such as using a diuretic, may be more appropriate. However, diuretics are commonly viewed as contraindicated because of the fear that they may depress RV function by unbalancing the RV preload (Frank Starling mechanism).
In a preliminary retrospective study, we tested the hypothesis that a single dose of diuretic treatment was safe and superior to fluid expansion in the setting of PE with RV failure.20 A total of 70 clinically stable patients were enrolled in this study. Thirty patients received fluid expansion (1.6±0.9 L of saline solution) and 40 received furosemide (78±42 mg) according to the choice of the physician in charge of the patient. Regarding safety, our results are reassuring. Indeed, in-hospital survival and PE-related shock were similar between the two groups. Regarding efficacy, all clinical parameters (including heart rate, systolic blood pressure and urine output, as well as respiratory parameters including SpO2 and need for oxygen support) significantly improved with diuretic treatment but not with fluid expansion. These results are encouraging but the retrospective design of the study carries all the limitations associated with that type of work. To confirm these results, a double-blind randomised study comparing furosemide treatment to placebo in the setting of normotensive acute PE with RV dilation will be performed.
Study design and objectives
The Diuretic versus placebo in Pulmonary Embolism with Right ventricular enlargement (DiPER) trial is a multicentre, prospective, randomised, placebo controlled and double-blind study that will assess the safety and efficacy of a single dose of furosemide as compared with placebo in normotensive patients with acute PE with RV dilation assessed by RV dilation on echocardiography or CT of the chest, and positive BNP (ClinicalTrials.gov identifier NCT02268903).
The primary objective of the DiPER trial is to demonstrate the 24 h clinical benefit of diuretic treatment with furosemide as compared with placebo. The secondary efficacy objectives are to assess the benefit of diuretic treatment on a major cardiac event, on duration of hospitalisation in intensive care unit (ICU), and on RV dilation and pressure overload assessed by echocardiography. Finally, the safety objective is to confirm the safety of diuretic treatment in acute PE with RV dilation. Furosemide will be tested against placebo because no benefit associated with fluid expansion has ever been demonstrated in PE. On the contrary, previous studies suggest that fluid expansion might have a deleterious impact on patients with RV enlargement.
Study population
The study population will consist of 270 patients who will be enrolled in 12 centres in France. Inclusion and exclusion criteria are detailed in table 1. Briefly, inclusion criteria include symptomatic acute PE with first symptoms within 15 days, associated with RV dilation on echocardiography or CT, or high BNP, and with one abnormal Pulmonary Embolism Severity Index (PESI) criteria among heart rate >10/min, systolic blood pressure <100 mm Hg and arterial oxyhaemoglobin level <90%. Main exclusion criteria are cardiogenic shock or hypotension, which will contraindicate diuretic treatment and require thrombolysis, and significant LV dysfunction, as it could induce a bias towards higher response to diuretic treatment.
Inclusion and exclusion criteria
RV/LV ratio has been chosen because of its association with poor outcome in PE.21 Moreover, in a previous study by Mercat et al,16 fluid expansion has been proven to be only efficient in patients with moderate RV dilation. Finally, in our preliminary report, furosemide infusion provided higher improvement in patients with RV/LV ratio ≥1.20 Regarding biological markers of RV dilation, we chose BNP because this marker of ventricular dilation has been shown to have a prognostic predictive value independent from echocardiography in different studies.21 ,22
Inclusion and randomisation
Patients who meet all the inclusion criteria and none of the exclusion criteria, and signing their written informed consent, will be enrolled by a designated investigator from the centre. Patient inclusion will be performed online (secured internet protocol) using the Cleanweb software. The patient inclusion number will be allocated online by Cleanweb (format: 000-0000-XX: centre number/number of inclusion/first initial of the name and first name). Patients will be randomised in consecutive order of qualification. A random allocation list will be drawn up by the clinical research unit statistician (URC-CHU Henri Mondor) before beginning the study. The treatment will be labelled and each treatment will be identified by a treatment number. The treatment number and random number will be the same. Randomisation will be carried out in blocks through a dedicated web-based system, which will also be used for electronic data capture during the study. Treatments, made available for each centre and stored in the investigator department, will be allocated by the investigator. Randomisation will have to be carried out within 4 h following confirmation of RV dilation and BNP measurement, and within 24 h following the diagnosis of PE.
Procedures
The study flow diagram is displayed in figure 1. After randomisation, a number corresponding to a single-dose vial containing the study drug or placebo will be assigned to the patient. Each vial will contain either furosemide 80 mg or placebo. Only the treatment number will be mentioned on the vial, so patients, investigators and anyone involved in care or in data analysis will be blinded to the treatment. Treatment will be administrated by a single intravenous injection (bolus injection) immediately after randomisation. Patients and investigators will both be blind regarding study treatment. The anticoagulant regimen will consist of unfractionated heparin, low molecular weight heparin or fondaparinux, according to local practice. Fluid expansion will be authorised if judged necessary, but will be recorded. Oxygen administration will be decided by the investigator and will be recorded. As per guidelines, thrombolytic treatment will not be allowed until clinical signs of shock, cardiac arrest or sustained hypotension occur. The dose of 80 mg for furosemide treatment was derived from the preliminary study, in which patients received 78±42 mg furosemide. We have demonstrated safety and potential efficacy with the dosage, therefore we chose it for the randomised study.

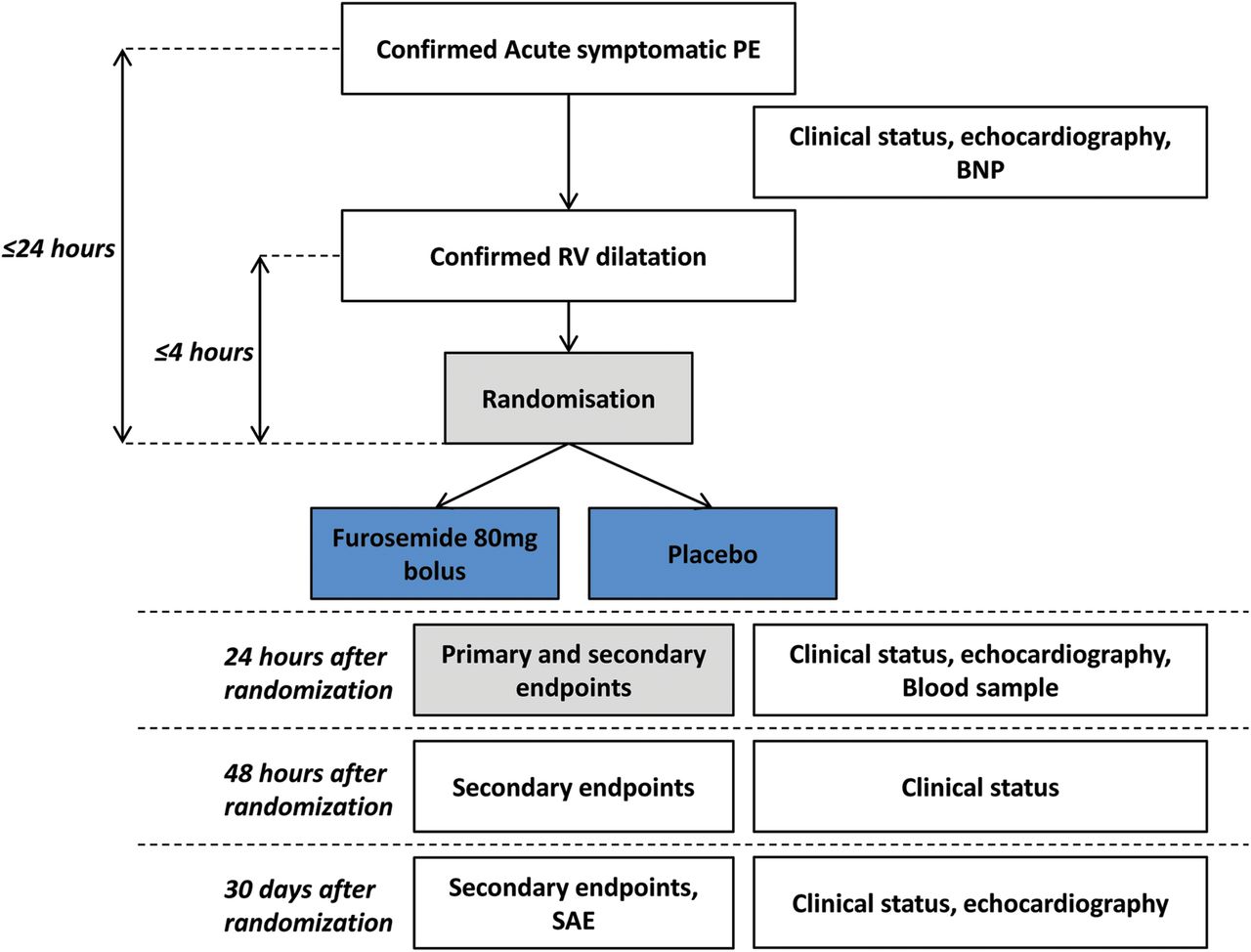{kind=link}
Flow diagram of the DiPER trial. DiPER, Diuretic versus placebo in Pulmonary Embolism with Right ventricular enlargement; PE, pulmonary embolism; BNP, brain natriuretic peptide; RV, right ventricle; SAE, serious adverse events.
Outcomes
The primary efficacy end point will be assessed at 24 h and will combine four clinical parameters derived from the PESI. To reach the primary end point, patients will have to meet all the following: (1) urine output >0.5 mL/kg/min for the past 24 h and normalisation of the clinical parameters of the simplified PESI (sPESI) score; (2) heart rate <110 bpm; (3) systolic blood pressure >100 mm Hg and (4) arterial oxyhaemoglobin level >90%.
Secondary efficacy end points will be the following: (1) major adverse event defined as death, cardiac arrest, mechanical ventilation, need for catecholamine and thrombolysis during hospitalisation; and (2) at 30 days; (3) duration of hospitalisation in ICU, (4) New York Health Association (NYHA) status at 1 month; (5) RV/LV ratio decrease (diameters and surfaces) from baseline to 24 h and 1 month; and (6) systolic pulmonary pressure decrease from baseline to 24 h and from baseline to 1 month.
Secondary safety end points will be the following: (1) major adverse event at 30 days (also an efficacy end point); (2) use of fluid expansion and (3) creatinine variations from baseline to 24 h.
sPESI estimates the risk of death or major complication after PE. sPESI includes age, underlying cancer, previous cardiorespiratory disease, heart rate, SpO2 and blood pressure. Thus, heart rate <110 bpm, systolic blood pressure >100 mm Hg and SpO2 >90% are associated with low risk of death or PE-related adverse event.23 We believe that urine output is another haemodynamic marker that reflects haemodynamic stability in patients with RV failure. The combination of urine output and sPESI clinical parameters is relevant as it indicates the disappearance of preshock symptoms and is therefore associated with a lower event risk. Thus, it allows early discharge of patients from the ICU. Other sPESI components (ie, history of cancer and age) were not included because these parameters cannot be influenced by any treatment strategy. Our preliminary study showed early improvement (24 h) of these parameters compared with fluid expansion. Therefore, we decided to evaluate the primary end point 24 h after treatment administration. Major cardiac event, duration of hospitalisation in ICU, and RV dilation and pressure overload improvement, will be evaluated as secondary end points.
Collection of data
Clinical parameters will be recorded. To optimise data quality measurement, the following instructions will be followed: systolic blood pressure will be calculated using the mean of three consecutive measurements, heart rate will be recorded over a whole minute, and arterial oxyhaemoglobin signal quality will be checked before recording and will be recorded without oxygen treatment for ≥5 min. For transthoracic echography, diameters and surfaces of left and right ventricles will be recorded in four-chamber apical view. Measurements will be performed just before QRS complex (ie, end-diastolic volumes). Pulmonary pressure measurement will be performed using tricuspid regurgitation velocity recorded using continuous Doppler. Right atrial pressure will be estimated using inferior vena cava diameter and variation. All echographies will be stored on DVD (DICOM) and analysed centrally by an independent physician who will be unaware of study treatment allocation (Henri Mondor Hospital).
To ensure accurate collection of the data, electronic case report form (eCRF) is made with range check for data values. Monitoring will be regularly performed. During these visits, the following elements will be reviewed: protection and safety, compliance with the protocol of the research, the procedures defined and regulations in force, quality of data collected in case report forms (accuracy, missing data, data consistency with the source documents (medical records, appointment books, original laboratory results, etc)). This aims to ensure the quality of research, the validity of its results, and compliance with the law and regulations in force.
Monitoring
A data monitoring committee (DMC) has been set up to assess the safety data of the trial and to provide an oversight over efficacy data after interim analysis. The DMC will provide recommendations to the sponsor as to whether the study should continue without change, be modified or be terminated. The DMC consists of two clinician expert of the disease, one pharmacologist expert and one methodologist expert. None of these members have any conflict of interest, and the DMC is independent of the sponsor.
Adverse events
All serious adverse events should be reported immediately to the sponsor. These include death, life-threatening events, events that require hospitalisation or prolongation of existing hospitalisation, and persistent or significant disability/incapacity. All other adverse events will be collected in the ‘adverse event’ section of the CRF.
Statistical analysis
The study is a prospective, multicentre, randomised (1:1), double-blind and parallel group comparison. The objective is to assess the benefit of furosemide between the two treatment arms at 24 h. Analysis will be conducted according to a prespecified analysis plan. All tests will be two-sided and p values less than 0.025 will be considered statistically significant due to an interim analysis that will be performed after recruitment of 50% of the population. All patients will be analysed in the group to which they were randomly assigned, according to the intention-to-treat principle. The primary outcome is binary: success if urine output is >0.5 mL/kg/h during the first 24 hours and normalisation of the three clinical parameters of the sPESI score (heart rate <110/min, systolic blood pressure >100 mm Hg and arterial oxyhaemoglobin level >90% on room air), and failure otherwise. Patients deceased before 24 h will be considered as failures. The null hypothesis is that there will be no difference between furosemide and placebo treatment. The alternative hypothesis is that furosemide will be superior over placebo on 24 h clinical improvement. The success rates will be compared between groups using the χ2 of Fisher's exact test, the relative risk and absolute risk reduction will be estimated with their 95% CI in order to estimate the effect size. Multilevel logistic models will be used to test a potential effect of the centres (stratification factor), the level 1 is the patient who is ‘nested’ in the level 2, the centre. In case of unbalanced characteristics, multivariate models adjusted for baseline characteristics will be performed. The same analyses will be performed for categorical variables; the Mann-Whitney U test will be used for continuous variables.
Censoring variables will be analysed using a survival model. The delay is defined as the time between the randomisation and the first event (death, thrombolysis, need for catecholamine, cardiac arrest and mechanical ventilation). Patients lost to follow-up and those who did not experience one of the events will be censored. Event-free survival will be estimated using the Kaplan-Meier method and treatment arms will be compared using the log-rank test. A multivariate Cox model will be performed. Variables will be selected on the p value in univariate analysis and then the model with the lower AIC will be selected. The stratification factor used in the randomisation will be included in the analysis. The proportional hazard assumption will be checked. In a second analysis, death will be considered as a competing risk. Adverse events (need for catecholamine, cardiac arrest and mechanical ventilation) will be represented as cumulative incidence. Factors associated with survival will be tested using a univariate Gray test, followed by a multivariate Fine and Gray model. For continuous end points evaluated at different time points, a linear mixed model for repeated measures will be undertaken. These models will be adjusted on the potential confounding factors. Assumption of the model will be checked.
Sample size/power calculation
The number of patients to be included was estimated to respond to the main primary end point, that is, the 24 h success rate. To potentially reduce trial duration and to avoid allocation of patients to an inferior treatment, an interim analysis will be performed. Therefore, a sample size adjustment was performed and a two-sided α risk of 0.025 has been considered. We previously observed a 46% success rate (95% CI 28% to 64%) among patients treated with fluid expansion and an 83% success rate among patients with furosemide (95% CI 71% to 95%). We postulated that the success rate in the placebo group will be close to the rate previously observed in patients treated with fluid expansion and, above all, lower than the upper limit of the CI (64%). Based on these two rates (83% vs 64%, 30% of relative difference), a two-sided α risk of 0.025 (1 interim analysis) and a β risk of 10%, 256 patients (128×2) are needed. Considering 5% of patients not analysable, 270 patients will be included (no or few lost to follow-up considering the 24 h assessment). Given the short duration of intervention (1 dose of treatment), it is highly unlikely that participants will discontinue or deviate from intervention protocol.
Discussion
The DiPER study is the first randomised study to introduce diuretic treatment in the setting of acute PE with RV enlargement. We hypothesise that furosemide may prevent patients with intermediate risk PE from developing cardiogenic shock and, furthermore, that furosemide may obtain faster improvement in these patients compared with placebo. Thus, it may allow early discharge of patients from the ICU. This study has not only been designed to confirm the haemodynamic improvement associated with diuretic treatment in PE with RV dilation as a primary end point, but also to assess if this haemodynamic improvement is associated with clinical benefit (secondary outcome). Moreover, repeated echographies will enable investigation of the mechanisms of haemodynamic improvement following diuretic treatment.
Patients with intermediate-risk PE as defined in the protocol represent about 20% of patients with PE admitted in cardiology wards. The simplicity of the protocol, number of recruiting centres and relatively small number of patients being included should easily enable enrolment of the required number of patients. Moreover, the protocol benefits from extensive experience gained through the PEITHO trial, which has recruited well and achieved high rates of follow-up.24
The main limitation of this study is that clinical benefit will not be assessed as the primary outcome. Therefore, the power of this study may not be adequate to show a clinical benefit.
References
Footnotes
Contributors RG, GM, JT, CB, AB, NM, RR, FC, MS, NL, HB, PC, BM, SB-G, J-LD-R and PL were involved in design of the study and protocol submission. RG, GM, JT, BM, SB-G, J-LD-R and PL were involved in redaction of the manuscript. CB, AB, NM, RR, FC, MS, NL, HB, PC and BM were involved in revision of the manuscript.
Funding The DiPER trial is registered with the ClinicalTrials.gov identifier NCT02268903. The trial is sponsored by public funding, specifically, by grants from the French government (Ministry of Health) (AOM 130519).
Competing interests None declared.
Ethics approval Comite de protection des personne Ile de France VII.
Provenance and peer review Not commissioned; peer reviewed for ethical and funding approval prior to submission.