Article Text

Abstract
Objectives This study aimed to detect α- and β-thalassaemia mutations in the Jino ethnic minority population of Yunnan Province, Southwest China.
Design A total of 1613 Jino adults were continuously recruited from February 2012 to April 2012. Fasting venous blood samples were obtained to determine haematological variables. Haemoglobin analysis was conducted using high-performance liquid chromatography. Participants with hypochromic microcytic anaemia or positive haemoglobin analysis profiles were confirmed by α- and β-globin genetic testing, including DNA microarray analysis, direct sequencing methods and multiplex gap-PCR assays.
Setting Shanghai Diabetes Institute, Shanghai Key Laboratory of Diabetes Mellitus, Shanghai Jiao Tong University Affiliated Sixth People's Hospital.
Results We found 363 suspected cases by primary screening of haematological variables and haemoglobin analysis. After further genetic testing, four types of α- and β-thalassaemia mutation were detected in 203 out of 363 individuals. Both α0- and α+-thalassaemia mutations, --SEA and -α3.7, were identified. β-Thalassaemia mutations included CD17 (HBB:c.52A>T) and CD26 (HbE or HBB:c.79G>A). In addition, 13 HbE carriers had coexisting α0- or α+-thalassaemia deletions. Clinical haematological variables indicated that, in this study, carriers of all thalassaemic genotypes had more severe hypochromic microcytic anaemia than non-thalassaemic individuals.
Conclusions Our results provide information on the Jino ethnic minority that may be useful for further genetic counselling, prenatal screening and clinical diagnosis of thalassaemia in this region.
- EPIDEMIOLOGY
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Strengths and limitations of this study
As Jino, the last ethnic minority confirmed in China, was reported to have a high prevalence of thalassaemia according to previous research on children under 10 years of age, this study aimed to detect mutations of α- and β-thalassaemia in Jino adults.
The α- and β-thalassaemia mutation spectrum shown in this research may help to explain further genotype–phenotype correlations and to establish a thalassaemia-prevention programme in this area.
The sample size we used in genetic testing was relatively small and may not have the validity to identify the rare thalassaemias in this ethnic group.
Introduction
As a group of monogenic disorders, thalassaemia is a serious health problem worldwide, especially in Mediterranean areas, Southeast Asia and Southern China.1–3 Yunnan Province, which is located along the border areas of China–Myanmar–Laos, is notable for its ethnic diversity. According to a previous study of children under 10 years of age, several ethnic minorities in this region have a high prevalence of thalassaemia, with the prevalence of α-thalassaemia (α-thal) being highest (22.1%) in Dai from Xishuangbanna and the prevalence of β-thalassaemia (β-thal) being highest in Achang (40.6%).4
Jino is the last ethnic minority confirmed in China, and the prevalence of α-thal and β-thal among Jino children are 3.1% and 29.3%, respectively. Thalassaemic children may exhibit various clinical symptoms; some are asymptomatic carriers, whereas others have severe haemolytic anaemia.5 Blood transfusion therapy, which is needed for severe carriers, imposes a heavy burden on families and public health management.6 Although genetic screening is essential to prevent and control this inherited disease, systematic investigations of thalassaemia mutations in Jino adults are rare.
The Jino population comprises nearly 20 000 individuals, and most (∼90%) live around Jino Mountain, which is located in East–Central Yunnan Province.7 A large number of thalassaemic mutations have been found in the general population worldwide;8–10 however, little is known about this isolated population. Indeed, the molecular mechanism and genetic variations of thalassaemia in Jino individuals may be different from those in other ethnicities. Our study aimed to detect α-thal and β-thal gene mutations in Jino adults to provide basic information for further prenatal consulting and thalassaemia diagnosis.
Materials and methods
Participants and clinical screening
Ethics approval for the study was granted according to the Declaration of Helsinki (paragraph II) by the institutional review board of Shanghai Jiao Tong University affiliated with the Sixth People's Hospital, Shanghai, China. This cross-sectional study was conducted between February 2012 and April 2012 in eight villages (Luote, Jiama, Balai, Situ, New Situ, Baka, Baya, Dapingzhang) around Jino Mountain in Jinghong, Southern Yunnan Province, China (figure 1). A list of Jino adults from these eight villages was obtained from local villager committee offices. Participants were sampled by a simple computer programme of randomisation from these villages. Staff at the local health centre, who understand both Chinese and Jino languages, contacted the subjects and introduced the purpose of the study. Oral and written informed consent was obtained from all the individuals. Basic demographic information and fasting venous blood samples were collected by researchers.

Geographical location of the Jino ethnic minority populations in Yunnan Province, southwest China. The solid black triangle represents Jino Mountain. The solid black circles represent the eight villages in Jinghong where the 1613 subjects were randomly selected.
A total of 1613 Jino adults, including 762 men and 851 women, participated in this survey (haematological and demographic characteristics of the total population included in the study are given in table 1). The following haematological variables were measured: haemoglobin (Hb), mean corpuscular volume (MCV), mean corpuscular haemoglobin (MCH), red blood cell (RBC) and red cell distribution width (RDW). Haemoglobin was analysed by high-performance liquid chromatography (HPLC) using the Variant II haemoglobin analysing system (Bio-Rad Laboratories, Hercules, California, USA). α- and β-globin genetic testing was performed in participants (n=363) with hypochromic microcytic anaemia (MCV <80fL and/or MCH <27 pg) and/or positive HPLC profiles. In order to evaluate the validity of primary screening approaches for detection of thalassaemia carriers, we randomly selected some of the individuals with negative screening results (n=50) from the remaining participants (n=1250) for further genetic testing.
Haematological and demographic characteristics of 1613 Jino ethnic minority adults included in the study
Genetic testing
Genomic DNA was extracted from venous blood leucocytes. Three methods were used to detect thalassaemic mutations.
A CapitalBio Thalassaemia Gene Mutation Detection Kit (CapitalBio, Beijing, China) was used to determine 25 common mutations in globin genes in the Chinese population via DNA microarray. Six α-thal gene mutations and 19 β-thal gene mutations were included. Among them, there were three α-thal deletions—that is, the Southeast Asian deletion (--SEA), rightward deletion (-α3.7) and leftward deletion (-α4.2)—and three non-deletional α-thal mutations—that is, Hb Constant Spring (HBA2:c.427T>C), Hb Quong Sze (HBA2:c.377T>C or HBA1) and Hb Westmead (HBA2:c.369C>G). Nineteen β-thal gene mutations were CD14/15 (HBB:c.45_46insG), CD27/28 (HBB:c.84_85insC), CD41/42 (HBB:c.126_129delTCTT), CD71/72 (HBB:c.216_217insA), -32 (HBB:c.-82T>C), -30 (HBB:c.-80T>C), -29 (HBB:c.-79A>G), -28 (HBB:c.-78A>G), CD17 (HBB:c.52A>T), CD26 (HBB:c.79G>A), CD30 (HBB:c.91A>G), CD37 (HBB:c.113G>A), CD43 (HBB:c.130G>T), IVS1-1 (HBB:c.92+1G>T), IVS1-5 (HBB:c.92+5G>T), IVS2-5 (HBB:c.315+5G>C), IVS2-654 (HBB:c.316-197C>T), Int (HBB:c.2T>G), CAP (HBB:c.-11_-8delAAAC). A BioMixer II Microarray Hybridisation Station (CapitalBio) was used for hybridisation after multiplex PCR amplification. Then, chips were scanned using a LuxScan 10K-B Microarray Scanner (CapitalBio).
To validate β-thal mutations, three fragments of the β-globin gene were amplified. The first fragment was amplified with 5′-CCT AAG CCA GTG CCA GAA GAG C-3′ as the forward primer and 5′-TGC CCA GTT TCT ATT GGT CTC C-3′ as the reverse primer, the second fragment was amplified with 5′-TAG AAA CTG GGC ATG TGG AG-3′ as the forward primer and 5′-TGT ACC CTG TTA CTT ATC CC-3′ as the reverse primer, and the third fragment was amplified with 5′-TCA GGG CAA TAA TGA TAC AA-3′ as the forward primer and 5′-TTA GTA GTT GGA CTT AGG GA-3′ as the reverse primer. The fragments were sequenced using a 3500 Genetic Analyser (Applied Biosystems, Foster City, California, USA).
We also confirmed three α-thal deletions—that is, the Southeast Asian deletion (--SEA), rightward deletion (-α3.7) and leftward deletion (-α4.2)—using multiplex gap-PCR assays. Primers and PCR conditions were designed as described in the literature.11 ,12
Statistical analysis
A statistical analysis was carried out using SAS for Windows (V.9.2). All quantitative traits were tested for normality, and skewed quantitative traits were logarithmically transformed to approximate univariate normality. Data are shown as means±SD. Quantitative traits (RBC, Hb, MCV, MCH, RDW) were compared between two groups using the Wilcoxon test, and analysis of variance was performed to compare the differences in the three subgroups of thalassaemia carriers (α-Thal, β-Thal and αβ-Thal). Two-tailed statistical significance was considered at p<0.05.
Results
Mutations identified in Jino
Owing to mutations in different globin genes, we observed three groups of thalassaemic carriers, including individuals with only α-thal gene deletions or β-thal gene mutations and individuals with combined αβ-thalassaemia (αβ-thal) gene mutations. Four different thalassaemia mutations were detected in 203 individuals among 363 suspected cases. No mutations were observed in 50 individuals with negative primary screening results. Table 2 shows the allele frequency of α- and β-thal mutations found in our study.
Allele frequency of α- and β-thalassaemia mutations found in our study
Mutations in the α-thal gene
None of the three common non-deletional α+-thal mutations, Hb Constant Spring (HBA2:c.427T>C), Hb Quong Sze (HBA2:c.377T>C (or HBA1)) and Hb Westmead (HBA2:c.369C>G), were found in 203 participants with thalassaemia mutations. Forty-six of 203 participants carried α-thal deletions only; --SEA and -α3.7 were observed, accounting for 16.7% (34/203) and 5.9% (12/203) of the mutations, respectively. Among these individuals, we identified both α+/α and α+/α+ for -α3.7 and α0/α for --SEA (gel electrophoresis of PCR amplifying results are shown in figure 2). However, no -α4.2 deletion was observed.

Gel electrophoresis of PCR amplifying results in α-thal deletions. M, marker, 200 bp DNA Ladder; lane 1, rightward deletion (genotype of -α3.7/-α3.7); lanes 2 and 3, rightward deletion (genotype of -α3.7/αα); lanes 4 and 5, Southeast Asia deletion (genotype of --SEA/αα).
Mutations in the β-thal gene
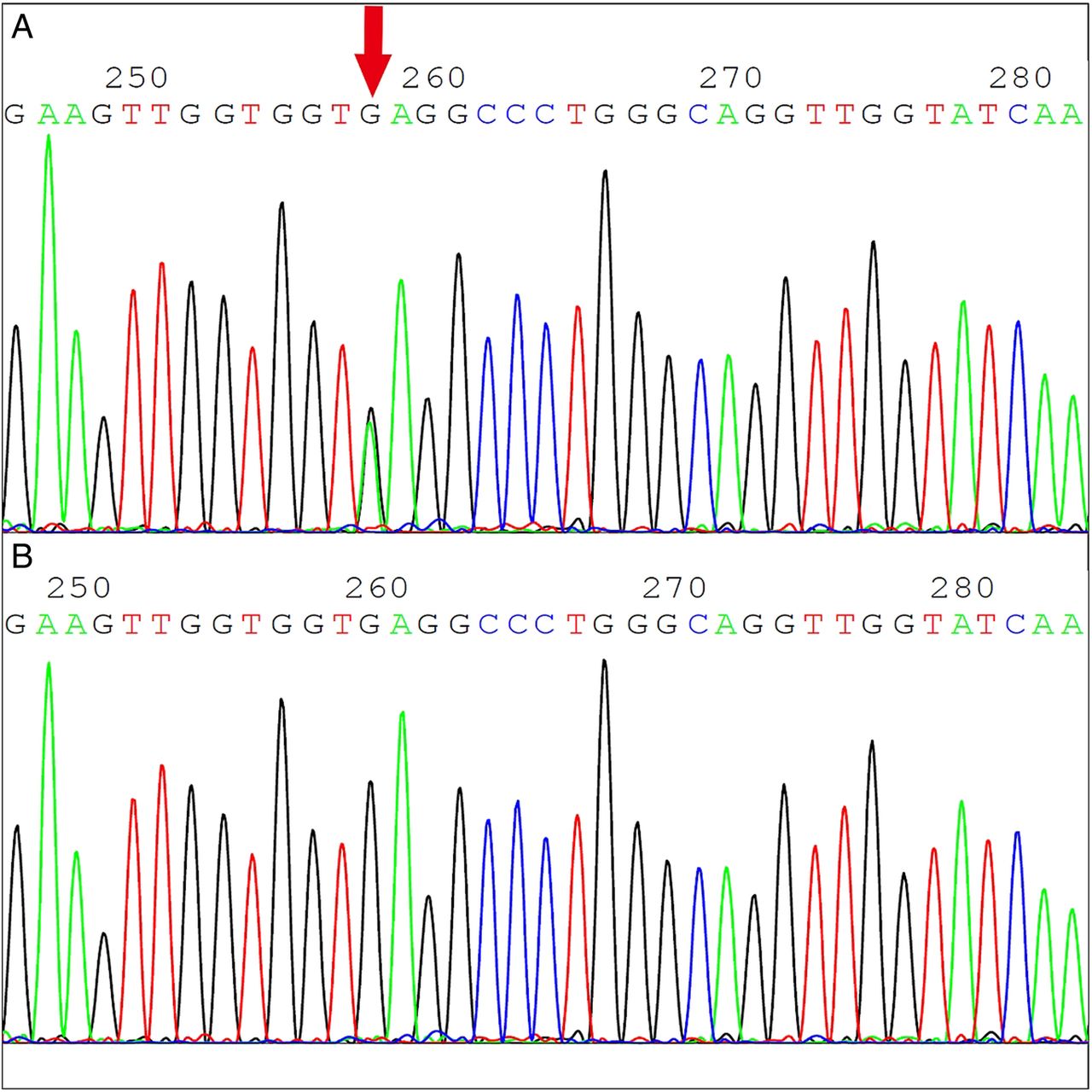
We observed mutations in CD17 (HBB:c.52A>T) (figure 3) and CD26 (HbE or HBB:c.79G>A) (figure 4). CD17, which accounted for 9.9% (20/203) of mutations, was found to be β0/βA in this population. Participants with HbE variant only, either βE/βA or βE/βE, accounted for 61.1% (124/203) of mutations. Furthermore, 13 HbE carriers harboured --SEA (n=8) or -α3.7 (n=5) at a combined frequency of 6.4% (13/203).

Heterozygous CD17 (A>T) mutation (A) and the corresponding normal sequence of β-globin. Red arrow indicates the position of this point mutation.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Heterozygous CD26 (G>A) mutation (A) and the corresponding normal sequence of β-globin. Red arrow indicates the position of this point mutation.
Haematological features of different thalassaemia genotypes
The haematological data of different thalassaemia genotypes are summarised in table 3. Compared with normal individuals, thalassaemic carriers had significantly lower Hb, MCV and MCH levels (p<0.001, respectively) and higher RBC and RDW levels (p<0.001, respectively). Furthermore, we compared the differences in these five indexes among the three groups of carriers (α-thal, β-thal and αβ-thal). Significant differences in MCV (p=0.0111), MCH (p=0.0002) and RBC (p=0.0012) were observed between these groups. MCV and MCH levels in the α-thal group were significantly lower than those in the β-thal group (p<0.05), whereas RBC levels were higher (p<0.05). In contrast, no difference was observed between the αβ-thal and α-thal groups/β-thal groups. Moreover, there was a tendency towards increased RDW levels in the α-thal group compared with the β-thal group (p=0.0573).
Haematological data of 1613 Jino ethnic minority individuals with different thalassaemia subtypes
Discussion
Thalassaemia is a common monogenic disease with a relatively high prevalence in Southeast Asia. In China, this disease is mainly prevalent in areas near the southern bank of the Yangtze River, such as Guangdong, Guangxi, Fujian and Yunnan Provinces.13–15 Prenatal screening and related molecular diagnoses are crucial for preventing and treating thalassaemia. Many thalassaemia studies have been conducted in Yunnan Province.16 ,17 However, data on the Jino population are limited because this population is the last ethnic minority confirmed in China.
We randomly selected 1613 Jino adults from eight villages around Jino Mountain in Jinghong, Southern Yunnan. Among the gene mutations identified, the most prevalent α-thal and β-thal genotypes in this region were --SEA and HbE, in agreement with previous data from Yunnan Province.18 ,19 According to our results, the overall prevalence of thalassaemia in Jino was nearly 12.6%, which is similar to the prevalence observed in Kunming.20 Prevalence of αβ-thal (8%) in our population was equal to that in the Li population in Hainan Province (7.99%), where thalassaemia prevalence is high.21 Although Yunnan Province has a high prevalence of thalassaemia with diverse genotypes, the spectrum of globin gene mutations among the Jino population is relatively limited.
HbE, a type of haemoglobinopathy, can be observed in most regions of Southeast China.22 Due to a point mutation in the β-globin gene, the balance of various globin products is disrupted, leading to a structural haemoglobin variant. Although HbE carriers may only have slight anaemia, their offspring will exhibit severe clinical symptoms in the presence of other β-thal types.23 Therefore, potential HbE carriers should undergo genetic testing and prenatal counselling. HPLC is often used as an efficient primary screening method to detect abnormal Hb, as was carried out in this study and previous studies.24 ,25 In our study, 95.6% (131/137) of HbE carriers were identified by HPLC, and 13 of these individuals had concomitant α-thal deletions.
Different genotypes lead to different clinical phenotypes.26 We found that thalassaemic carriers had significantly lower MCV and MCH levels. Regarding those with β-thal mutations, MCV and MCH levels were significantly decreased in CD17 carriers compared with HbE carriers, suggesting that a nonsense mutation in the β-globin gene causes greater erythrocyte impairment. Hypochromic microcytic anaemia was moderate in individuals with βE/βA and α+/α compared with βE/βA carriers. This paradox may be explained by the fact that changes in the α- and β-globin chains may balance each other out when both mutations coexist in an individual. Accordingly, rapidly estimating the genetic state of an illness based on haematological variables is difficult. Therefore, genetic screening of both α- and β-globin gene mutations in potential parents is of utmost importance to prevent births with severe defects.27
However, there are some limitations in this study. First, the sample size we used in the genetic testing was relatively small and may not have the validity to identify the rare thalassaemia variants from this ethnic group. Second, investigations of population and family structure were not performed in this study, although α-thal and β-thal gene mutations were common among the Jino ethnic minority. As a result, further studies on the inbreeding levels and consanguinity structure are warranted to reveal the underlying mechanism of gene flow and then assess the occurrence and persistence of α-thal and β-thal gene mutations, especially coexisting α-thal and β-thal gene mutations within Jino individuals.
In conclusion, this study revealed α- and β-thal mutations in the Jino ethnic minority population in Yunnan Province. Of these mutations, --SEA and HbE were the most prevalent α-thal and β-thal gene mutation types, respectively. In addition, data based on clinical haematological variable analysis indicated that the severity of hypochromic microcytic anaemia is associated with the genotype of thalassaemia. Our results provide evidence that may be useful for further genetic counselling, prenatal screening and clinical diagnosis of thalassaemia in this region.
Acknowledgments
We thank all of the participants for their dedication. We acknowledge the skilful technical support of all nursing and medical staff at the Shanghai Clinical Center for Diabetes.
References
Footnotes
Contributors WJ and CH conceived and designed the experiments. SW and RZ performed the experiments and analysed the data. GX, YL, XH, FuJ and FeJ contributed materials and analysis tools. SW prepared the article. CH and WJ revised the manuscript. All the authors have read and approved the final version of this manuscript.
Funding This work was financially supported by a grant from the National Natural Science Foundation of China (91331110).
Competing interests None declared.
Patient consent Obtained.
Ethics approval According to the Declaration of Helsinki (paragraph II), ethics approval for the study was granted by the institutional review board of Shanghai Jiao Tong University affiliated with the Sixth People's Hospital, Shanghai, China. Written and Oral informed consent was obtained from all participants.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement No additional data are available.