Article Text

Abstract
Introduction This study aims to explore the feasibility of using moxibustion as a supplementary intervention and to assess the sample size for verifying the effectiveness and safety of integrative treatment involving moxibustion compared with conventional treatment for patients with benign prostatic hyperplasia accompanying moderate to severe lower urinary tract symptoms.
Methods and analysis A total of 60 patients diagnosed with benign prostatic hyperplasia by a urologist based on prostate size, prostate-specific antigen and clinical symptoms will participate of their own free will; urologists will monitor the patients and evaluate their symptoms. The patients will be randomised to either a conventional group or an integrative group with a 1:1 allocation according to computer-generated random numbers concealed in opaque, sealed, sequentially numbered envelopes. Watchful waiting or oral medication including α blocker, 5α-reductase inhibitors or antimuscarinic drugs will be offered as conventional treatment. Integrative treatment will include moxibustion therapy in addition to the conventional treatment. The moxibustion therapy will be conducted twice a week for 4 weeks on the bilateral acupoints SP6, LR3 and CV4 by a qualified Korean medical doctor. The primary outcome will be the International Prostate Symptom Score (IPSS) after eight sessions. The secondary outcomes will be the post-void residual urine volume, the maximum urinary flow rate, IPSS, the results of a Short-Form 36-Question Health Survey after 12 weeks, and the patients’ global impression of changes at each visit.
Ethics and dissemination Written informed consent will be obtained from all participants. This study was approved by the institutional review boards of both Pusan National University Yangsan Hospital and Pusan National University Korean Medicine Hospital. The trial results will be disseminated through open-access journals and conferences.
Trial registration number NCT02051036.
- moxibustion
- integrative medicine
- lower urinary tract symptoms
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Strengths and limitations of this study
The design of this clinical trial is based on a conference of experts, including Korean medical doctors (KMDs), urological doctors (UDs) and an Eastern–Western integrative medicine specialist who has both MD and KMD licenses, to develop an optimal integrative treatment.
Optimal conventional oral medications and a customised number of moxibustion layers for each patient are used to reflect the real clinical setting.
This study's results can serve as a basis for further large studies or studies of intractable urinary disorders.
The statistical power of the study may be low because of the small sample size.
Practitioners and patients will not be blinded.
Introduction
Korean statistical data show that the prevalence of benign prostatic hyperplasia (BPH) in men over 65 years was 17.9% in 2011,1 and BPH ranked 25th among male outpatient visits by frequency of disease in 2013.2
BPH causes lower urinary tract symptoms (LUTSs) by directly disturbing the bladder outlet or increasing the tension and resistance of smooth muscles.3 For treatment, watchful waiting at the beginning and behaviour modification with oral medication are recommended,3 ,4 and these methods have proved effective in improving LUTSs, urinary flow rate and post-void residual urine in many previous studies.5–7
However, this conventional treatment is limited by certain side effects. For α blockers, rhinitis (6.6%), dizziness (4.4%) and abnormal ejaculation (2.8%)5 caused by tamsulosin have been observed. Moreover, abnormal ejaculation (14.2–28.1%) caused by silodosin,8 ,9 cardiovascular adverse events (5.7% hypertension, 3.9% non-hypertension)10 and mild dizziness (13.9%)11 caused by alfuzosin, severe dizziness leading to drug suspension (2.0%) caused by terazosin,12 and dizziness (4.41%), postural hypotension (4.03%) and asthenia (4.08%)13 caused by doxazosin have been verified. In addition, erectile dysfunction (4.53%), decreased libido (2.36%) and abnormal ejaculation (1.78%)13 caused by finasteride have been identified, and dry mouth (24%), dyspepsia (5%), back pain (5%) and micturition disorder (5%)14 caused by tolterodine have been shown to occur. In particular, when two or more types of these medications are combined, each side effect is expected; thus, careful use only for patients with moderate to severe BPH is recommended.4
To overcome this limitation, many studies investigating complementary and alternative medical (CAM) treatment have been conducted, but the 2011 American Urological Association's (AUA's) guidelines reported that no definite evidence exists to recommend CAM treatment because of the lack of quality and quantity of CAM studies of BPH.3
In contrast, clinical studies of acupuncture or herbal medication for BPH with LUTSs have been consistently performed15–19 and have demonstrated the effectiveness of these methods. Moxibustion has been shown to be effective in treating urinary disorders,20 but well-designed clinical trials to prove its effectiveness are lacking. Therefore, we designed a pilot trial to explore the feasibility of moxibustion as an adjuvant for BPH with LUTSs based on its effectiveness and safety and to estimate the appropriate sample size for a future, large comparative effectiveness study, with the purpose of developing an optimal integrative treatment acceptable to both medical doctors (MDs) and Korean medical doctors (KMDs) in the present medical system. The design of this clinical trial is based on a literature survey and a conference of experts including KMDs, urological doctors (UDs), and an Eastern–Western integrative medicine specialist who has both MD and KMD licenses. This pilot study is a randomised controlled trial with a parallel-group, 1:1 allocation, exploratory and pragmatic design.
Methods and analysis
Aims
The present study aims to evaluate the feasibility of moxibustion as an adjuvant for conventional treatment in patients with BPH and to determine the correct sample size for verifying, in future studies, the effectiveness and safety of the integrative treatment compared with conventional treatment for patients with BPH accompanying LUTSs. This is a single-centre, assessor- and analyser-blinded, parallel-group, 1:1 allocation, pragmatic randomised controlled study.
Recruitment
Notices were posted in front of the Pusan National University Yangsan Hospital (PNUYH) urological office and the Pusan National University Korean Medicine Hospital (PNUKH) genitourinary clinic office, and an advertisement for the study was also placed on the internet homepage of PNUKH. A UD will confirm the diagnosis of BPH and the impracticality of surgical treatment for patients who volunteer to participate. A KMD will thoroughly examine all inclusion/exclusion criteria and explain the trial to eligible patients. When the patient decides to participate in the study, the KMD will obtain written informed consent, and a baseline assessment will be performed. The study will consist of a screening phase, a treatment phase and follow-up. A more detailed description of the study is shown in figure 1. The time schedule for participation is shown in table 1.
Progression of trial

Trial flowchart. BPH, benign prostatic hyperplasia; IPSS, International Prostate Symptom Score; KMD, Korean medical doctor; PSA, prostate-specific antigen; TRUS, transrectal ultrasonography.
Study design
Randomisation and allocation concealment
Within 14 days of recruitment, each patient will be allotted to the conventional or integrative group according to the concealed random list. A statistician who is not taking part in the study will place the computer-generated random list into each double-layered opaque envelope, seal it and write the numbers in sequence. The KMD will give the envelope to the patient according to visit order and open it with the patient.
Patients
Sample size
The sample size was not calculated on the basis of a power calculation because this is a pilot study. It was determined from estimates of the number of patients expected to participate and the minimum number needed to evaluate the pragmatic purpose of the trial. Thus, a sample size of 30 per group and a total number of 60 will be included, which is larger than the minimum number recommended for pilot studies.21
Inclusion criteria
Male patients aged 20–80 years diagnosed with BPH with a prostate size over 20 g
International Prostate Symptom Score (IPSS) ≥8
Written informed consent obtained
Patient must be able to check symptom severity according to the IPSS
Exclusion criteria
Prostate or bladder malignancy
Received herbal medication for LUTSs within the last week
History of a brain disease that can cause urinary difficulty
Difficulty answering the IPSS because of cognitive impairment
Signs of acute urinary tract infection
Has diabetes mellitus
Neurogenic bladder
Drop-out criteria and process of management
Definition of drop-out cases
Completed cases will be defined as patients who finish the treatment process and follow-up. Patients who cannot complete the study because of side effects or for other reasons will be considered drop-out cases.
Drop-out criteria
The researcher may stop treatment and observation of a patient according to prescribed criteria, and the patient can drop out voluntarily at any time. The drop-out criteria are as follows.
Violation of inclusion/exclusion criteria
Serious adverse events or adverse events making a patient wish to drop out
Severe systemic disease that was not recognised at baseline
Patients or a legal representative demand cessation of the trial because of unsatisfactory effects or withdrawal of consent
Trial compliance of less than 80%; should attend at least seven of the eight treatment sessions in the integrative group and all of the three major assessments (baseline, visit 9 and visit 10) in both groups
Protocol violation of patient or researcher
Difficulty conducting moxibustion because of newly developed disease or uncooperative manner
Patients not replying to outcome measures
Patient's desire or UD's recommendation for surgical treatment (including minimally invasive therapies)
Management process
The drop-out date, time and reason will be recorded on the end report. Patients can drop out voluntarily for any reason, at any time, and are not required to submit a reason. The researcher should make every effort to follow-up patients who have dropped out and record the reason for drop-out or the reason for not being able to determine a drop-out cause.
Blinding
Blinding of outcome assessors and data analysers
The practitioner and patients cannot be blinded because this is an open-label study for moxibustion treatment. Assessors and analysers will be blinded. Urodynamic testing will be performed by an assistant who is not taking part in the trial, and participants will be asked not to reveal their allotted group to the assistant. Subjective outcomes will be recorded by the patient. The groups will be marked ‘A’ and ‘B’ when the data are sent to the statistician to ensure that the groups are not recognised as the control group and experimental group. Unblinding of the assessors will be permissible only in the case of a serious adverse event.
The rationale for the lack of a sham moxibustion group
For the pragmatic purpose of the future study, we decided to reflect the real clinical situation without omitting the patients’ additional time, effort and expectations by comparing patients who receive conventional treatment with patients who receive both conventional treatment and complementary treatment. A sham or placebo intervention group is the ideal method for efficacy studies with an optimal, strictly restricted design to minimise all influencing factors to prove the efficacy of a specific component of the intervention.22 ,23 Consequently, a sham moxibustion group will not be included in this study.
Interventions
Conventional treatment protocol
The conventional treatment will be set as the optimum treatment for each patient to develop a reasonable integrative treatment protocol.24
The optimum treatment for each patient will be based on the UD's opinion. The UD will discuss behavioural modifications, such as water intake, with the patient. Watchful waiting will be used for patients without renal insufficiency, urinary retention, recurring infection or complications of bladder outlet obstruction. Oral medications will be prescribed when no therapeutic effect is observed after watchful waiting, and the medications will be selected taking into consideration overactive bladder, prostate size and prostate-specific antigen (PSA). Preferentially, an α blocker, such as alfuzosin, doxazosin, tamsulosin or terazosin, will be used for functional symptom mitigation. 5α-Reductase inhibitors, such as dutasteride and finasteride, will be used when the prostatic volume is >40 mL or PSA >1.4 ng/mL. In cases of a high risk of BPH progression, prostate ≥30 mg or PSA ≥1.5 ng/mL, a combination of an α blocker and a 5α-reductase inhibitor will be used. Anticholinergic agents, such as tolterodine, will be prescribed for patients with overactive bladder but will need to be monitored for patients with ≥250 mL post-void residual urine. The conventional treatment components can be changed at the discretion of the UD because this research is a pragmatic study to evaluate the effectiveness and safety of additional moxibustion therapy, and the conventional treatment will be maintained for the last follow-up.3 ,4
Integrative treatment protocol
Moxibustion therapy will be added to the conventional treatment described in the section ‘Conventional treatment protocol’ twice a week for 4 weeks. The moxibustion therapy will be conducted by a skilled KMD with at least 2 years’ experience in the clinic. The timeframes of the conventional treatment group and the integrative treatment group are shown in figure 2. Both apparatus-type and mini-pillar-type moxibustion will be used.

Timeframe of the integrative treatment group (I.G) and conventional treatment group (C.G).
The apparatus types are a Hatnim-moxa (Bosungsa, Incheon, South Korea) and a moxa pillar (Bosungsa), which generates 65–70°C of heat. Moxibustion will be conducted at bilateral acupoints CV4 on one layer of gauze (figure 3) for 30 min. This acupoint was selected on the basis of KM theory20 ,25 and previous clinical studies.17 ,26 Additional gauze will be offered layer by layer when the patient requests it because of intolerable heat. This apparatus-type moxibustion will be stopped if the patient still complains of intolerable heat even after additional gauze has been offered more than three times. If the patient cannot feel any heat once the moxa pillar is totally burned, the moxibustion will be conducted one more time; the procedure can be performed once more as before, but the number of moxibustion applications cannot exceed three. The therapy can be stopped if a second-degree or higher burn occurs before completion of the eight treatment sessions.

Apparatus-type moxibustion on acupoint CV4.
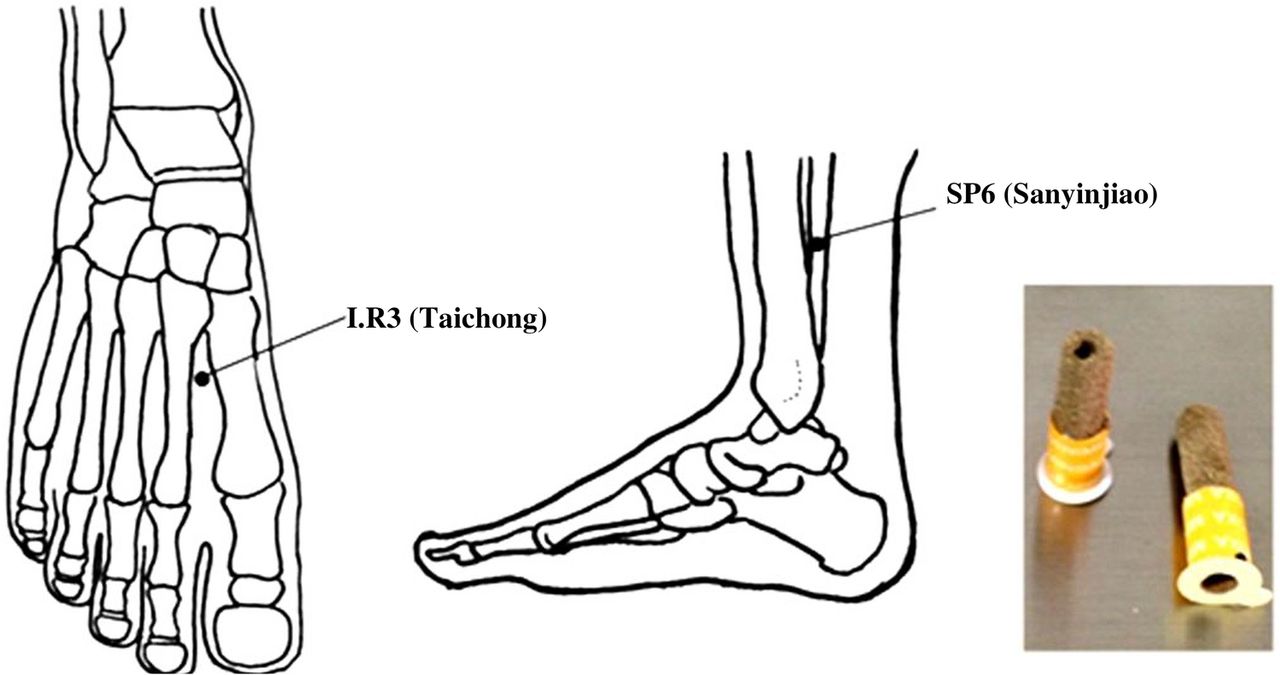
Kanghwa mini-moxa of ‘lowest’ intensity, which generates heat of approximately 45°C, will be used for mini-pillar-type indirect moxibustion. Mini-moxa will be conducted at bilateral acupoints SP617 ,20 ,27 and LR320 ,28 ,29 (figure 4). The mini-moxa will be removed when totally burned, which takes approximately 5 min, but it may be removed if the patient complains of intolerable heat. The mini-moxa will not be repeated on the acupoint on which moxibustion was stopped at the patient's request. Repetitive mini-moxa will be performed on the acupoints at which the patient did not feel heat, and completed before mini-moxa for the entire burning period, up to a maximum of seven times on each point. Beginning with the second session, the treated region will be checked, and further mini-moxa will not be allowed on an acupoint where a second-degree or higher burn occurred. In this case, the mini-moxa will be restarted after the burn is completely healed.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Mini-pillar-type moxibustion on bilateral acupoints SP6 and LR3.
The treatment session of twice a week for 4 weeks was determined based on the studies of Yang et al,30 Liu et al31 and Wang et al,16 who reported the effective results and clinical experiences of two KMDs considering practicality in terms of the patients’ general social environment and the accessibility of the hospital. The number of moxibustion treatments performed will be recorded in both the electronic medical record and the case report form (CRF) at every visit in adherence with intervention protocols.
It is reasonable to stop moxibustion therapy if a second-degree or higher burn occurs according to KM theory,32 and, in such a case, the patient will not be considered a study drop-out. However, if a patient cannot continue the treatment because of discomfort from the moxibustion smoke, allergic response or pigmentation from the moxa-soot, he/she cannot be regarded as having completed the trial.
Prohibited or allowed parallel medical treatments
Flexible oral medications according to the discretion of the UD will be allowed to offer the best treatment for each patient; thus, the medications will not be fixed without variation. All types of medication therapy based on AUA and Korean Prostate Society guidelines will be allowed.
Surgical treatment, including transurethral resection of the prostate, transurethral incision of the prostate, abdominal prostatectomy, minimally invasive therapy using a laser, transurethral needle ablation of the prostate and transurethral microwave thermotherapy, is prohibited; therefore, patients who want, or are recommended for, such therapies cannot participate in this trial.
Treatment of adverse events
We will disinfect and dress the wound when a second-degree or higher burn occurs and will refer the patient to dermatology to receive proper treatment when a third-degree or higher burn occurs.
Outcome measures
Primary outcome measure
The IPSS after eight sessions will be the primary outcome measure. The results of the IPSS, including the change in IPSS and its SD, will be used to calculate the proper sample size for the future trial by performing a power analysis. IPSS was developed by the AUA in 1992, and a question regarding quality of life (QoL) was later added.33 The Korean version was validated in 1996.34 IPSS consists of seven sub-themes: incomplete emptying, frequency, intermittency, urgency, weak stream, straining and nocturia. The severity scoring is as follows: 0–7, mildly symptomatic; 8–19, moderately symptomatic; 20–35, severely symptomatic. The separate QoL question requires the respondent to select a QoL category ranging from 0 (delighted) to 6 (terrible) (table 2).
International Prostate Symptom Score
Secondary outcome measures
Patient's global impression of changes (PGIC)
PGIC will be recorded for each patient at every visit after the first treatment. PGIC is a scoring system used to evaluate the level of change from the beginning of the treatment, either conventional or integrative, to the time of the PGIC check. This scale considers limitations of physical activity, symptoms, emotions and QoL in general. The scoring is as follows: no change (or condition has become worse), 1; almost the same, hardly any change at all, 2; slightly better, but no noticeable change, 3; somewhat better, but the change has not made any real difference, 4; moderately better and a slight but noticeable change, 5; better and a definite improvement that has made a real and worthwhile difference, 6; a great deal better and a considerable improvement that has made a substantial difference, 7. In a similar way, each patient will be asked to circle one of the numbers 0 (much better) to 10 (much worse) written on a straight line that represents the change from the beginning to the time of evaluation.35
The Short-Form 36-Question Health Survey (SF-36)
The SF-36 will be checked at the baseline, after four sessions, after eight sessions and after 12 weeks from the baseline. The SF-36 is a commonly used scale to evaluate health-related quality of life. This scale consists of physical function, physical role capability, bodily pain, general health perceptions, vitality, social role capability, emotional role capability and mental health.36
The maximum urinary flow rate (Qmax)
An independent tester will measure Qmax at baseline and 12 weeks after baseline. Qmax changes from baseline to 12 weeks will be used as an objective outcome measure. Urodynamic study is an invasive method to obtain objective and quantitative data on bladder outlet function and storage function. Patients will attend the study when they feel a ‘normal’ desire to urinate. The velocity of the external urine stream will be automatically obtained by a calculation using the voided volume and time.37
Post-void residual urine volume (PVR)
An independent tester will measure PVR at baseline and 12 weeks after baseline. PVR changes from baseline to 12 weeks will be evaluated because PVR increases when bladder outlet function is incomplete.37 PVR will be checked by ultrasonography immediately after the urodynamic study.
Changing progress and persistency on IPSS
The IPSS will be evaluated after four sessions in the integrative group to explore the process of change and will be checked 12 weeks after the beginning of the study, after 8 weeks of completed moxibustion therapy, to evaluate the persistency of the effects of moxibustion therapy. This period of 12 weeks was determined on the basis of a previous study,26 the clinical experiences of two KMDs, and the optimum follow-up period recommended in conventional treatment guidelines.38–40
Adverse events
To explore safety, adverse events will be recorded. At every visit, patients will be asked whether adverse effects have developed and, if so, what types of adverse effects. In particular, second-degree or higher burns and allergic responses of the skin or whole body will be examined thoroughly, and other types of discomfort will be checked.
Recruitment, compliance and retention rates
For feasibility, the recruitment, compliance and retention rates will be recorded. The recruitment rate will be the ratio of the patients who completely meet the inclusion/exclusion criteria and who register for the trial versus the recruitment goal. The compliance rate will be measured by the attendance rate for the treatment phase in the integrative group and the attendance rate for the three major assessments (baseline, visit 9 and visit 10) in both groups. The retention rate is defined as the ratio of (1) the number of patients who attend the primary outcome assessment after 4 weeks versus the total number of participants, (2) the number of patients who attend the final assessment after 12 weeks versus the total number of participants, and (3) the number of patients who return the frequency–volume chart versus the total number of participants.
Data collection
Subjective outcome measurements will be checked for each patient, and objective outcome measurement data will be preserved in both their original form and as an electronic medical record. These data will be written on the CRF by a certificated clinical research coordinator. To promote patient retention and completion of follow-up, an honorarium will be provided with a differential rate according to the patients’ participation.
Statistical analysis
Analysis of efficacy
Both intention-to-treat and per-protocol analyses will be performed. The last-observation-carried-forward method will be used for missing data in intention-to-treat analysis. The paired t test will be used for intragroup before/after treatment comparisons. The independent t test will be used for intergroup comparisons. For non-parametric data, the Wilcoxon signed-rank test for intragroup and the Wilcoxon rank-sum test for intergroup test will be used. Categorical data, such as adverse effects, will be investigated by calculating the occurrence rate of adverse events for each group and then performing analysis with the χ2 test or Fisher's exact test. If statistically significant differences between two groups are observed or covariance is expected, analysis of covariance will be used. All of the statistical analysis will be performed with two-tailed tests, and the significance level will be set as 0.05. To explore feasibility, the recruitment, compliance and retention rates will be calculated, and the percentages will be reported. Furthermore, subgroup analyses will be performed according to the severity in terms of the IPSS or prostate size and the type of conventional treatment.
Safety
Expected adverse events, such as burns and allergic responses, will be recorded along with their modality, date of occurrence, and duration. Patients will report other unexpected adverse events freely. The severity of the adverse events will be categorised according to the WHO 5-grade performance status classification as follows: 0, able to carry out all normal activity without restriction; 1, restricted in strenuous activity but ambulatory and able to carry out light work; 2, ambulatory and capable of all self-care but unable to carry out any work activities, and up and about more than 50% of waking hours; 3, symptomatic and in a chair or in bed for more than 50% of the day but not bedridden; 4, completely disabled, unable to carry out any self-care; 5, totally confined to bed or chair. The cause-and-effect relation between the intervention and adverse events will be assessed according to the WHO-Uppsala Monitoring Centre (UMC) causality categories: 1, certain; 2, probable/likely; 3, possible; 4, unlikely; 5, conditional/unclassified; 6, unassessable/unclassifiable.
To minimise the expected adverse events, we will describe the risk of adverse events to patients who have had prior allergic responses to moxibustion therapy, allergic rhinitis or allergic conjunctivitis. Patients will be told to notify the practitioner if they experience such symptoms during the treatment so that they can receive proper and prompt treatment. The treatment will be performed in a well-ventilated room, and patients will be offered a mask to cover their mouth and nose. To prevent burns, patients will be educated about indirect moxibustion therapy and its precautions and told to notify the practitioner promptly if they feel intolerable heat and wish to stop the treatment. The principal investigator (PI) will describe and assess all of the symptoms that occur during the clinical trial and will report to the institutional review board (IRB) to determine whether to continue or stop the study when serious adverse events occur.
Patients who suffer from adverse events will be treated as described in the section ‘Treatment of adverse events’. In addition, patients who suffer harm from participation in this trial will be cared for through insurance. All patients will be informed of, and sign off on, the ‘regulation concerning subject compensation’, including detailed descriptions of this regulation.
Monitoring
The independent data monitoring committee (DMC), composed of one KMD and one clinical research expert, will examine the process of progress and whether the trial follows the study plan, the standard guidelines and clinical trials management criteria and other related standards. Monitoring will be conducted by regular visits and phone calls. The DMC will check the original record and CRFs. If any problem is found, the DMC will discuss this with the PI. If any serious problems that could threaten the safety of patients are found, the DMC will discuss this with the IRB and PI. The PI will make the final decision as to whether to continue or to terminate the trial, and the IRB can order the PI to terminate the trial in the case of a serious problem.
Ethical considerations and dissemination
Written informed consent and study approval
This study was approved by the IRBs of both PNUYH and PNUKH. Signed informed consent will be given by each patient to the practitioner. If any changes to the inclusion/exclusion criteria, outcome measure methods or data analysis are demanded, the decision will be made through a discussion between the UD and KMD. Any changes in content will need to be reapproved by the IRB and reflected in the patient explanation and study registration (clinicaltrials.gov), and fresh consent from the patient will need to be obtained.
Private information protection
Collected data from patients will be safeguarded with specific serial numbers without any personally identifiable information so that nobody will be able to recognise the patients except a security manager who has a code table. Computer-stored personal information will be secured using a password, and all matters related to security will be supervised by the PI. Publication will not include any personally identifiable information, and data will be treated anonymously. Strict security is assured even in the case of a patient dropping out mid-study and after the end of the study.
Data used for the study will be disposed of after the collection of material for a research paper. Computer storage files will be deleted and documents will be shredded on 31 November 2017.
The PI, a monitor and an inspector can read the patients’ records for the purposes of monitoring and progress oversight in terms of laws and ethics. These data will be stored securely in the National Clinical Research Center for Korean Medicine. This matter will be explained to the patients, who will also be provided with a written explanation.
Dissemination
The trial results will be disseminated through open-access journals and conferences.
Discussion
This study is the first protocol of a randomised controlled pilot trial in Korea to evaluate the feasibility of moxibustion as an adjuvant to conventional therapy for BPH accompanying LUTSs by exploring its effectiveness and safety.
The medical system of South Korea has been maintained as a dualised system since the revival of KMD by the enactment of the National Medical Insurance Act in 1951. This system has had some negative aspects, such as the incautious use of medicine combinations and distrust between the two medical fields; however, it has had some positive aspects as well, including providing patients with a large variety of treatment choices.41 Therefore, the need for integrative medicine has been propounded steadily to establish a new medical system combining the advantages of Western and Korean medicine.42
This study was designed as an investigation of add-on treatment without a placebo control because additional alternative treatments in conjunction with the conventional treatment are considered appropriate in light of the medical ethics and medical treatment system.23 ,43 Despite relatively acceptable rates of adverse events, increased side effects caused by the combination of different types of oral drug4 and by patient vulnerability factors, such as ageing and underlying disease, must still be investigated. Therefore, the effectiveness and safety of adjuvant treatments should be evaluated, after which the adjuvant treatment may be considered for intractable urinary disorders including interstitial cystitis and chronic prostatitis. In addition, a pragmatic design is used to improve applicability to the clinical field and decision-making.44 ,45 Thus, we set broad inclusion/exclusion criteria and flexible interventions allowing for different treatment regimens according to each patient's medical condition. In addition, considering the pragmatic purpose and the study ethics, conventional oral medication is not restricted to one type. Moxibustion therapy has the limitation of inconvenience because patients must visit the hospital for every treatment, whereas conventional oral medication can be provided once for a relatively long period; thus, after discussion, the experts decided to perform treatment for a relatively short period of time and to follow-up after 12 weeks, as in conventional treatment.38
This study has some limitations. The 12-week follow-up was set according to the routine check period of conventional treatment, but this was not sufficient to evaluate the long-term effects. Furthermore, development of changes cannot be investigated because frequent and regular IPSS checks were not planned in this trial. Therefore, future trials should include more frequent and regular outcome assessments in both groups and a longer follow-up period of at least 1 year in order to investigate development of changes in each group and persisting effects, so that the treatment sessions, period and interval can be appropriately modified for the final integrative treatment guideline. Another limitation is that prostate size is not included as an outcome measure because the feasibility of the treatment is the main focus rather than definitive assessment of its effectiveness. Therefore, studies evaluating the effect of the combined treatment on prostate size should be conducted after the LUTS-reducing effect is demonstrated. Future power analysis studies should be performed by determining the effect size based on the results of this study, and cost-effectiveness studies should be performed to provide important information for decision-makers.
Trial status
This study is currently in the recruiting phase. The first patient was enrolled on 10 March 2014, data collection will be complete in approximately December 2015, and the article including results is expected in approximately 2016.
Acknowledgments
This manuscript was edited by American Journal Experts.
References
Footnotes
Contributors J-NK, S-DL and J-KN conceived the study. J-NK, S-DL, J-KN, J-YH, Y-JY and H-YL initiated the study design, and D-HL, S-HP, J-HL and H-LP helped with its implementation. J-KN, D-HL, J-HL and H-YL performed the intervention and discussed the optimal complementary medicine. H-YL drafted the study protocol manuscript. All authors contributed to the refinement of the study protocol and approved the final manuscript.
Funding This study was supported by a Grant from the Korea Institute of Oriental Medicine (K15042).
Competing interests None declared.
Patient consent Obtained.
Ethics approval The study was approved by IRBs of both Pusan National University Korean Medicine Hospital (IRB approval number 2013021) and Pusan National University Yangsan Hospital (IRB approval number 03-2013-013).
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement The data from this trial will be accessible by contacting the corresponding author. The trial results will be disseminated through open-access journals and conferences.