Article Text

Abstract
Introduction Verrucae are extremely common, and are experienced by most people at some time during their lives. Although most verrucae will spontaneously disappear without treatment, many patients seek treatment, often because they have persisted for many years, are unsightly or painful or prevent them from doing sports or other activities. There are many different treatments available; including the Falknor's needling procedure. To date, there has only been one small trial evaluating the clinical effectiveness of this treatment and no health economic analysis has been undertaken. The Effective Verruca Treatments (EVerT2) trial aims to evaluate the clinical and cost-effectiveness of the needling procedure for the treatment of verrucae.
Methods and analysis This single-centre randomised controlled trial will recruit 58 participants (aged 18 years and over with a plantar verruca) from Salford Podiatry Clinic patient lists and the surrounding area. If the participant presents with multiple verrucae, an ‘index’ verruca (largest and thickest lesion) will be identified and patients will be randomised 1:1 to the intervention group to receive the needling treatment or the control group to have the callus overlying the verruca debrided. The primary outcome is complete clearance of the index verruca at 12 weeks after randomisation. Secondary outcomes include clearance and recurrence of the treated verruca, clearance of all verrucae, number of verrucae remaining, change in size of the index verruca, pain, and participant satisfaction. A cost-effectiveness analysis of the needling versus callus debridement will be carried out from the perspective of health services over a time horizon of 12 weeks.
Ethics and dissemination Ethical approval has been obtained from the University of Salford, Department of Health Sciences Ethical Approval Committee (HSCR15/24) and the University of York, Department of Health Sciences Research Governance Committee (HSRGC/2014/98/B). Findings will be disseminated through publication and conference presentations.
Trial registration number ISRCTN16429440.
- plantar warts
- needling
- callus decridement
- verrucae
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Strengths and limitations of this study
The design of the trial is robust. The randomisation process and concealment of allocation are adequate, with blinded primary outcome assessment.
This study is a single-centre study in a University Podiatry clinic and the intervention will be delivered by up to two podiatrists. Although this ensures standardisation of the intervention protocol, the results may not be generalisable to other settings or to patients with other types of cutaneous warts.
The trial is powered to show a clinically meaningful difference in clearance rates of verrucae of 40 percentage points (from 30% in the control group to 70% in the intervention group) at 12 weeks after randomisation.
Introduction
Verrucae (or plantar warts) are caused by the human papilloma virus.1 They are common, and can present in people of all ages. Population-based studies estimating the prevalence rates of verrucae have produced varying results, ranging from 0.84% (USA),2 to 3.3% (UK),3 and up to 12.9% (Russia).4 Verrucae can be asymptomatic, unsightly, painful and debilitating. Although some verrucae will spontaneously disappear without treatment,5 some verrucae remain for many years, motivating patients to seek treatment.
The rate of natural clearance of warts from initial infection is not clear. Early research by Massing and Epstein5 suggests that there is a greater chance of clearance within the first 2 years of infection. However, clearance rates reported in the placebo arms of trials suggest that clearance may take place sooner than this. A Cochrane systematic review of wart treatments reviewed 21 trials with placebo groups,6 and found a clearance that averaged 27% (range 0–73%) in the placebo groups after an average period of 15 weeks (range 4–24 weeks). These data have led some practitioners to recommend that warts should not be treated at all.1 ,7 However, there is no reliable means of predicting which warts will clear spontaneously and which will remain for years.
The Cochrane systematic review assessed the effectiveness of different local treatments of cutaneous non-genital warts.8 The report highlighted the considerable uncertainty around the optimal treatment of verrucae. In 1969, Falknor9 described a needling technique for the treatment of verrucae. The technique involves administering a local anaesthetic to the region where the verruca is present. A needle is then inserted into the verruca until it exits the verruca tissue and enters the underlying dermis and subcutaneous fat layer. Once the boundary between the verruca capsule and the underlying fat is pierced, the needle is removed and the same process is repeated in an area of tissue immediately adjacent to the previous puncture until the entire surface of the verruca has been punctured. It is hypothesised that the mechanical trauma to the viral tissue may enhance the inflammatory response and hence the immune response within the area.10 In a retrospective review of 45 patients who underwent the needling procedure, 31 (69%) cases demonstrated complete clearance of verrucae, 3 (7%) demonstrated a reduction in size and pain and 11 showed no improvement 8 weeks after treatment. No adverse events were reported from any of the patients.11 To date, we are aware of only one trial evaluating the effectiveness of this needling procedure.12 This was a small trial involving 37 participants, randomised to receive either the needling technique or cryotherapy. Results of this study showed a statistically significant difference in the clearance of the primary pedal wart in the needling group of 64.7% (11/17) compared with 6.2% (1/16) in the cryotherapy group (p=0 .001) 12 weeks after the initial treatment. Unlike the retrospective study,11 this study involved delivering the treatment up to three times at least 2 weeks apart. There was no significant difference in pain or satisfaction with treatment between the two groups.
Objective
The objective of the EVerT2 (Effective Verruca Treatments) trial is to evaluate the clinical and cost-effectiveness of the needling procedure compared with callus debridement for the treatment of verrucae. The null hypothesis is that there is no difference in the clearance rates of the index verruca as assessed by a blinded independent assessor at 12 weeks postrandomisation between the two treatment groups.
EVerT2 is the second randomised controlled trial (RCT) conducted in collaboration with the York Trials Unit (YTU) at the University of York, focusing on the treatment of verrucae. The first trial (EVerT) tested the efficacy of salicylic acid and cryotherapy treatments for verrucae.13
Methods and analysis
Trial design
This will be a single-centre, pragmatic, open, two-arm RCT with an economic evaluation. Participants with verrucae will be randomised 1:1 to receive the needling procedure or to callus debridement. Participant progress through the study is shown in figure 1.
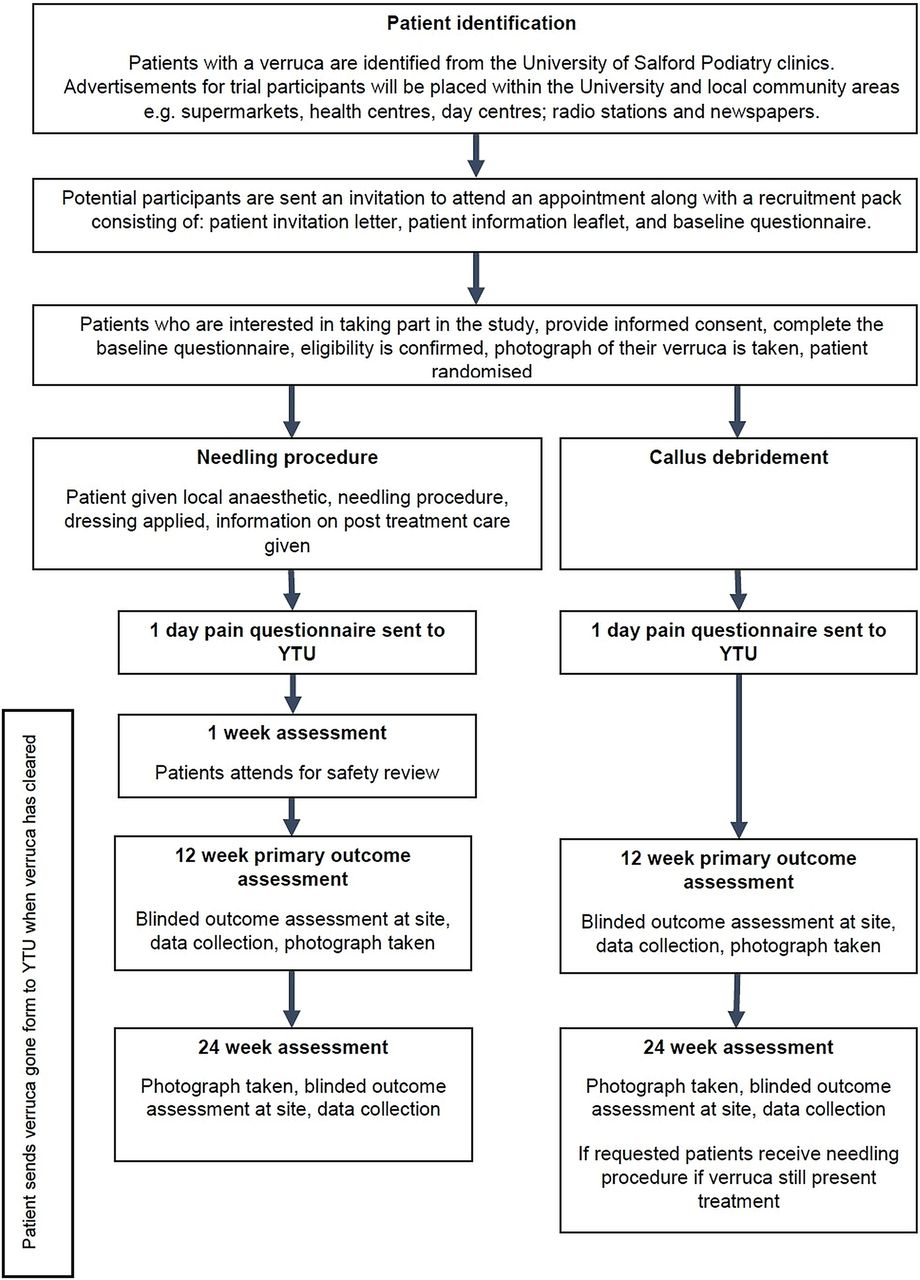
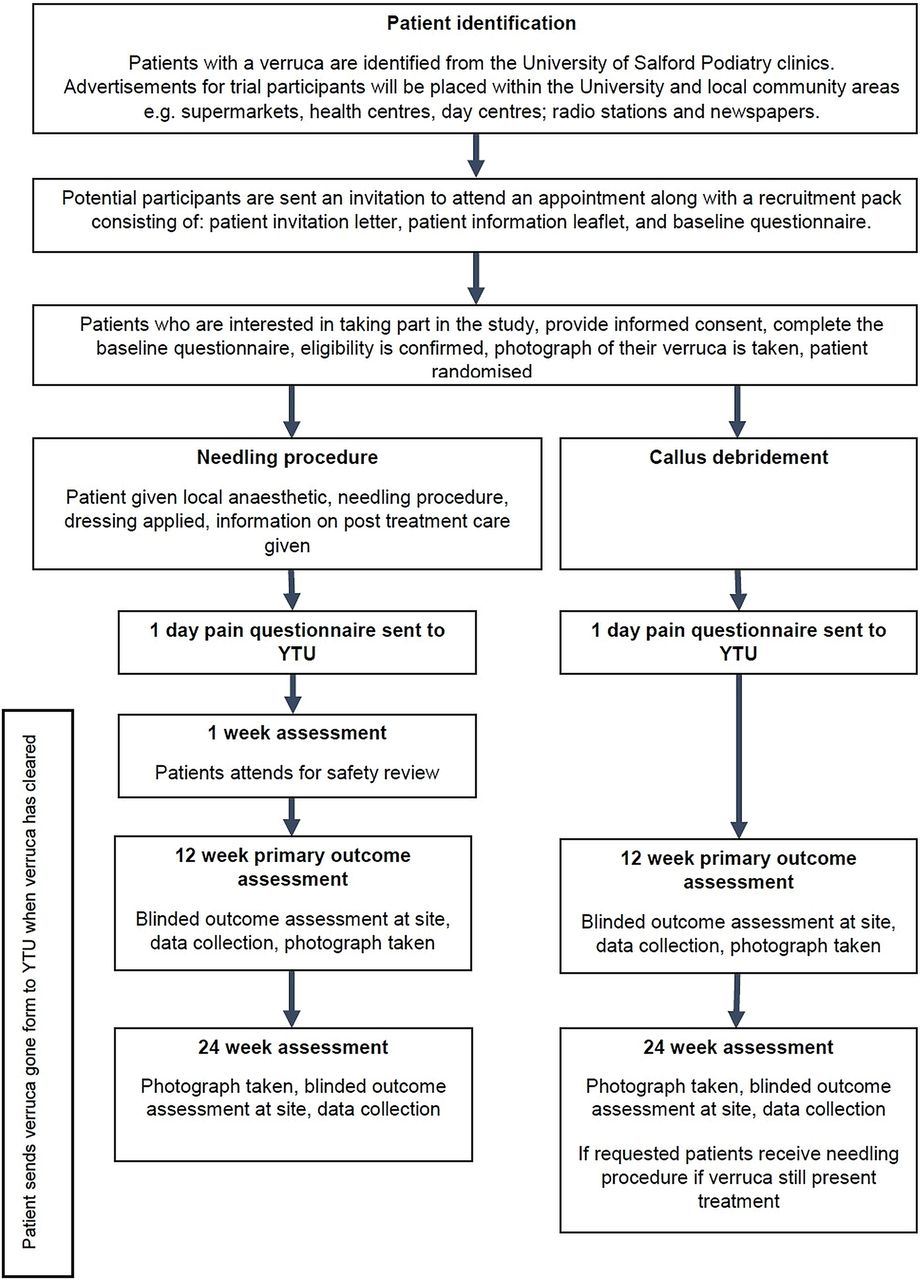{kind=link}
Flow chart of participants through the trail (YTU, York Trials Unit).
Setting and participants
The trial will be conducted in the Podiatry Clinic at the School of Health and Social Care, University of Salford, UK. Data management and statistical analysis will take place at the YTU, University of York, UK.
Recruitment
The podiatry clinic at the University of Salford is a private, teaching clinic. A self-referral system is in place for all patients. Potential participants for the trial will be identified by a qualified podiatrist, registered with the Health Care Professions Council (HCPC; UK) at the study site. In order to facilitate recruitment the following strategies will be used:
The podiatry clinic has a range of patients in the system with verrucae. These participants will be viewed on the computer system and those deemed to be suitable (ie, fit and healthy, and aged 18 or over) will be sent a letter advertising the study and asking them to contact the investigators if they are interested in participating.
Flyers will be distributed within the University, including the podiatry clinics, the Student's Union, the leisure centre and University campus buildings.
Recruitment flyers will also be distributed in local community areas accessed by the general public including supermarkets, health centres, day centres, leisure centres and athletics clubs.
Radio and/or newspaper advertisements will be used to give a brief description of the project and invite individuals that are interested in taking part to phone or email for further details.
Adverts may be placed on the University of Salford's Twitter feeds; other twitter or other social media accounts used for professional scientific purposes; or other electronic noticeboards within the University of Salford.
Potential participants will be sent an invitation letter, information sheet about the trial and a baseline questionnaire and will be provided with an appointment for assessment/treatment. For individuals responding to an advert for trial participants, telephone screening by the study site will be undertaken to ensure that the potential participants fulfil the inclusion criteria. Participants will be given a minimum of 24 h to read the information sheet and consider participation. At the baseline appointment, the podiatrist will ensure that the patient is eligible for the trial, take consent, check that all baseline data have been completed and randomise the patient. The patient will then receive the allocated treatment at the same appointment. Participants’ general practitioners (GPs) will be notified of their involvement in the trial after recruitment.
Inclusion criteria
Potential participants will be eligible if they have a plantar verruca that, in the opinion of the podiatrist, is suitable for both treatments: scalpel debridement and needling. The clinical diagnosis of a verruca will be made by a qualified podiatrist, registered with the HCPC (UK), observing the following criteria: flesh coloured, hyperkeratotic lesion with discrete margins and a papillomatous surface. The skin striae will be either broken or diverge around the lesion. Some verrucae may contain black dots of thrombosed capillaries. The lesions are often painful when pinched. Verrucae on weight bearing skin sites only will be included. All participants will be aged 18 years and older.
Exclusion criteria
Potential participants will be excluded if, in the opinion of the podiatrist, they are not suitable for local anaesthesia. Local anaesthesia is an absolute contraindication in potential participants who have a history of type 1 hypersensitivity reaction to local anaesthesia; who have infection or poor tissue viability at the site of injection; who do not consent (refusal) or when there is non-cooperation on the participant's behalf (this includes the patient moving the foot when attempting to inject or any other circumstance when the patients actions put themselves or the practitioner at risk of needle stick injury). Relative contraindications include participants: in the first trimester of pregnancy; with compromised renal and hepatic function; with cardiorespiratory problems such as angina and myocardial infarction; who are elderly or debilitated patient; with marked peripheral neuropathy; with progressive neurological disease; with epilepsy still suffering seizures.
Exclusion will also apply if the potential participants: have impaired healing (eg, due to diabetes, peripheral vascular disease or any other condition); are immunosuppressed (or are currently taking immunosuppressant drugs such as oral corticosteroids); have peripheral neuropathy; are currently on renal dialysis; are pregnant; are unable or unwilling to give informed consent; or are currently in a trial evaluating other treatments for their verruca(e).
Sample size estimation
With regard to the efficacy of the needling procedure, a retrospective review of 45 patients who underwent this procedure found that 69% cases demonstrated complete clearance of the verrucae 8 weeks after treatment.11 A recent RCT compared the efficacy of two verrucae treatments (needling procedure and cryotherapy with liquid nitrogen).12 This study found a 65% clearance rate of the index verruca among the participants allocated to the needling procedure compared with 6% among those treated with cryotherapy after 12 weeks of follow-up.
The EVerT2 trial is powered to show a more conservative but still clinically meaningful difference in clearance rates of the index verruca of 40 percentage points (from 30% in the control group to 70% in the intervention group) at 12 weeks after randomisation. A Cochrane systematic review of wart treatments found that the placebo arms of 21 trials had an average clearance rate of 27% after an average period of 15 weeks; hence, a control rate of 30% was chosen. In addition, powering to detect a difference from 70% to 30% requires the largest sample size to detect a 40 percentage point difference. For 80% power (5% two-sided significance), 26 participants per group are required. Allowing for 10% attrition, we will require 58 participants to be recruited and randomised (29 into each treatment group).
Randomisation
The random allocation sequence will be generated by the trial statistician using block randomisation with randomly permuted block sizes. Allocations will be concealed in sequentially numbered, opaque, sealed envelopes kept at the YTU. The allocations will be concealed from the podiatrists enrolling the participants to the study. Once a patient is ready to be randomised, the podiatrist recruiting the patient will ring the secure randomisation telephone number at the YTU for the treatment allocation.
Interventions
Intervention group: needling procedure delivered by a HCPC registered podiatrist
If the participant presents with mosaic verrucae or multiple plantar warts, the index verruca (the largest and thickest lesion) will be selected for treatment. The area surrounding the lesion will first be cleansed with skin disinfectant. A local anaesthetic (Scandonest 3%) will be administered via tibial nerve block, digital block or local infiltration according to the location of the lesion chosen for needling. Once the area of skin is anaesthetised, any callus overlying the verrucae will be debrided.
An empty surgical needle (21 gauge) will be used to repeatedly puncture through the lesion to the subcutaneous tissue. Each puncture will produce point bleeding and this will be continued until there is no more resistance, or reactive pressure, from the epidermis and the entire lesion has perforated enough to produce a ‘beefy’ red wound. The total number of punctures will vary according to the size of the lesion.
Pressure will then be applied to the wound with a sterile gauze. The wound will be dressed with a non-adherent sterile dressing (Melolin) and fixing tape (Mefix). A semicompressed felt aperture pad will also be applied to deflect pressure when walking and reduce postoperative bruising. Each participant will be issued with a postoperative care leaflet and advised to lightly shower and wash the area after keeping the dressing and padding dry for 24 h after treatment. Each participant will be advised to avoid taking non-steroidal anti-inflammatory drugs (such as ibuprofen) or other anti-inflammatory medication for 48 h to increase the likelihood of a successful controlled inflammatory response. Patients will be permitted to take paracetamol if they require pain relief.
This intervention will be delivered on one occasion only. Patients will attend a review appointment 1 week after the treatment where wound inspection and debridement of any uncomfortable eschar will be performed, and 12 weeks after randomisation for primary outcome data collection. At the 12-week appointment overlying callus will be debrided if the lesion is causing discomfort to the participant. Details of any treatment or advice given will be recorded on a podiatrist questionnaire. The final inspection for verrucae clearance will be carried out at 24 weeks postrandomisation. If the verruca has not cleared at this stage then further, alternative treatments may be offered.
Control group: debridement of callus overlying verruca delivered by a HCPC registered podiatrist
The area surrounding the lesion will first be cleansed with skin disinfectant. The callus overlying the verrucae will be removed using a surgical blade. This procedure is common practice for the removal of plantar callus. If the participant presents with mosaic verruca or multiple plantar warts, then all verruca callus will be debrided if the participant requests this and the podiatrist considers that it is safe to do so. However, the index verruca will be selected on which to assess outcome measures. Post-treatment, a semicompressed felt aperture pad will be applied to deflect pressure when walking and reduce postoperative bruising. Each participant will be advised to lightly shower and wash the area after keeping the dressing and padding dry for 24 h after treatment. Patients will attend a review appointment 12 weeks after randomisation for primary outcome data collection. If the patient is experiencing pain or discomfort due to the callus, then the callus overlying the verruca(e) will be further debrided. Details of any treatment or advice given will be recorded on a podiatrist questionnaire. The final inspection for verrucae clearance will be carried out at 24 weeks postrandomisation. If the verruca has not cleared at this stage, then further alternative treatments may be offered, which may include the needling procedure.
A ‘no treatment’ arm will not be included in this study. It is likely that people will volunteer for this study to access the novel treatment needling procedure which is not widely available either at the National Health Service (NHS) or private podiatry clinics. The risk of losing participants to follow-up would be high if these participants are randomised into a ‘no treatment’ group. Therefore, offering callus debridement, which is currently the mainstay of treatment in some NHS podiatry clinics, may help to maximise participant retention throughout the trial, particularly with the offer of free needling treatment at the end of the trial if their verrucae persist. Second, a ‘no treatment’ arm may lead to bias due to resentful demoralisation, particularly in those participants where the verrucae are painful, long standing and have been resistant to previous treatment. The use of a ‘sham’ needling treatment for the control group was considered; however, the trial team concluded that it would be unethical to administer a local anaesthetic if no treatment is to be given.
All treatments will be conducted by up to two podiatrists proficient in the needling procedure. In order to standardise understanding and implementation of study procedures, the podiatrists will review the following documentation together prior to the start of study: background and aims of the trial; inclusion/exclusion criteria; the randomisation process; protocols for both treatments; documentation/forms used in the trial, for example, randomisation forms, questionnaires, adverse event reporting; and possible ‘Frequently asked questions’ participants may ask about the trial.
All participants will receive a high street shopping voucher for the sum of £20 in recognition of their participation in the study and to offset any incidental expenses associated with completing the questionnaires and attending clinics. This money will be split between the 12-week and 24-week follow-up appointments.
Outcome measures
Baseline assessment
After written informed consent has been obtained, baseline data will be collected using a baseline questionnaire. The following data will be collected: participant gender and date of birth; the type (plantar calcaneus, plantar MTPJ, mosaic or other) and number of verruca(e); size of the index verruca; the length of time the participant has had the verruca(e); previous treatment for current verruca(e); reason for seeking treatment; level of pain; number of previous verrucae; and age of last verruca.
Primary outcome
The primary outcome will be complete clearance of the index verruca at 12 weeks after randomisation as determined by blinded assessment by a HCPC registered podiatrist working at the clinical site. ‘Clearance’ of verruca will be defined as the restoration of normal skin on close inspection, with the return of normal dermatoglyphics to the treated area of skin, that is, uninterrupted skin striae as assessed by the podiatrist. The blinded outcome assessment will be undertaken by a member of the research team who is unaware of the treatment the participant has received. The assessor will record whether or not the verruca has completely cleared on the podiatrist outcome assessment form. Participants will be reminded not to tell the person undertaking the blinded assessment which treatment they received.
Participants who do not attend their 12-week appointment will be contacted and asked to email a digital photograph of their verruca to the podiatry clinic email account with their participant ID number and the date the photo was taken.
Secondary outcomes
The secondary outcomes will be: pain 24 h after the treatment, clearance or recurrence of the treated verruca at 24 weeks, clearance of all verrucae, number of verrucae remaining, change in size of the index verruca, pain, and participant satisfaction at 12 and 24 weeks.
Data on self-reported clearance of verrucae, self-reported time to clearance, recurrence of verrucae at 24 weeks, satisfaction with treatment and pain levels will be recorded using patient questionnaires. Data on the number of treatments, number of verrucae remaining at 12 and 24 weeks, and change in size of verrucae at 12 and 24 weeks will be recorded by the treating podiatrist using a podiatrist questionnaire.
In addition to this, side effects of treatment, pain intensity after treatment and use of painkillers will be recorded and assessed by a participant questionnaire, which will be given to the participant after their first treatment. The participant will be asked to complete and send this questionnaire back to the YTU 1 day after their treatment, in the prepaid envelope provided. Participants will be given a ‘verruca gone’ form and stamped address envelope at their first treatment session, which the participant will complete and send back to the YTU when they assess their verrucae have gone.
Table 1 contains details of the outcomes that will be measured throughout the trial. The majority of the outcomes are responses to questions that require a ‘yes’ or ‘no’ answer or open comments, apart from measurements of verruca size; pain and satisfaction measures. Verruca size measurements will be taken from digital photographs using ImageJ software (V.1.49p, National Institute of Health, USA, http://imagej.nih.gov/ij, Java V.1.6.0_24 (32 bit)). A standard protocol will be used for all photographs, as follows. The participant trial ID card will be placed directly under the verruca, so that the scale is perpendicular to the base of the verruca. All attempts will be made to ensure that the ID card is not at an angle to the base of the verruca and is firmly adhered to the foot. A white card will be placed behind the foot before taking the photograph. Each photograph will be taken at a distance of 10 cm away from the foot. This is a sufficient distance to ensure that the verruca and the ID card are in the picture frame. All attempts will be made to ensure that the camera is not at an angle to the bottom of the foot. The flash facility will not be used as the research room has sufficient lighting. The zoom facility will not be used, that is, after turning on the camera the zoom will not be adjusted. The image will be opened in the ImageJ software package and the area of the lesion will be measured three times and the mean value used as the outcome measure. These measurements will be taken by a researcher who is blinded to the intervention. Pain will be measured using a 100 mm visual analogue scale. Participant satisfaction will involve asking the following question ‘How happy are you with your treatment?’ and recording the response using the following Likert scale: very unhappy, unhappy, neither happy nor unhappy, happy or very happy.
Details of outcome measures and data collection forms
Adverse events
The podiatrist will routinely record and report any serious adverse event (SAE) and non-SAE/reactions, which are directly related to the trial treatments or related to being in the study, which occur during the course of the trial. Any adverse events deemed not related to the trial treatment or related to being in the study will not be recorded or reported.
An adverse event is defined as any untoward medical occurrence which has occurred in a patient taking part in the study. A SAE is defined as any untoward occurrence that: results in death; is life threatening; requires hospitalisation or prolongation of existing hospitalisation; results in persistent or significant disability or incapacity; consists of a congenital anomaly or birth defect or is otherwise considered medically significant by the investigator.
Adverse events will be recorded on an adverse event form, as appropriate. An assessment of the seriousness, causality, expectedness and intensity of the event/reaction will be undertaken. Participants will be asked at each trial visit whether they have had any problems following their treatment and if any adverse events have occurred. The adverse event reaction reporting period for this trial begins when the participant is randomised into the study and ends 24 weeks after the date of randomisation. All recorded and reported adverse events and reactions will be followed up until they are resolved or the participant's participation in the trial ends. In addition, serious adverse reactions assessed by the investigator as being possibly related to the investigational procedure will continue to be followed even after the participant's participation in the trial is over. Such reactions will be followed until they resolve or until the investigator assesses them as ‘chronic’ or ‘stable’. Appropriate ongoing care will be arranged through the appropriate services. Resolution of such events will be documented on the adverse event form.
Potential complications associated with the needling procedure and callus debridement are: pain, irritation, infection, bleeding for longer than clinically expected following the needling procedure and scarring. In very rare cases patients may fall as a result of the treatment.
Data handling
Transfer of data and data entry
Data shall be collected via paper questionnaires. Where data collection is conducted at the University of Salford, completed paper questionnaires will be scanned and electronic copies will be sent to the YTU, University of York, via the University of York's secure, file exchange DropOff service. Wherever possible, identifiable details on any correspondence or records will be removed and replaced with the participant’s unique trial ID number. The YTU will not hold personal details of participants or consent forms. In cases where participants are asked to complete a questionnaire at home, a stamped addressed envelope for the YTU will be supplied.
Data from participant and podiatrist questionnaires returned to the YTU will be manually entered into bespoke databases, housed on the YTU secure server. The YTU has a backup procedure approved by auditors for disaster recovery. YTU-based standard operating procedures will be used for entering and checking data. Data quality checks will be undertaken to ensure the accuracy of the data. Any data queries will be raised with the site.
Data storage
All data will be treated with the strictest confidence and in accordance with the Data Protection Act 1998. Paper study documents will be retained in a secure (locked when not in use) cabinet in a locked room both during and after the trial has finished. During the course of the trial any personal identifiable paper records will be stored separately from anonymised paper records. Once the study is complete, data will be temporarily held in the department archive before being sent to the University of Salford for archiving. Data will be put in sealed boxes. Access to the archive is restricted and is via the named archivist. Access will only be allowed if a data query arises which requires access to the data in order to resolve the issue. All electronic records will be stored on a secure, password protected server within the YTU indefinitely.
Statistical analysis
There will be a single analysis at the end of the study using two-sided significance tests at the 5% significance level for the primary outcome measure and 1% significance level for secondary outcome measures. We will use ‘intention-to-treat’ analysis including all participants in the groups to which they were randomised irrespective of whether or not they received their allocated treatment. Analysis will be conducted in Stata V.13 or later (StataCorp LP, College Station, Texas, USA). Results will be reported according to CONSORT guidelines and a flow diagram depicting the progression of patients through the trial will be presented.
Trial completion and baseline data
Participants will have the option, at any time, of withdrawing from the trial treatment; withdrawing from completing postal questionnaires; and withdrawing from the collection of data by the podiatrist. If the participant elects to withdraw from all three, then he or she will be deemed as a full withdrawal. Healthcare professionals are able to indicate any change in the patient's level of participation by completing a change of circumstances form. The numbers of participants withdrawing from treatment and/or the trial will be summarised with the reasons where available.
Participants will be deemed to exit the trial when: the participant has been in the trial for 24 weeks; the participant wishes to withdraw from the trial fully; the participant's healthcare professional withdraws him/her from the trial; the participant is lost to follow-up; or the participant dies.
Baseline data
All data collected at baseline such as gender, age, type and duration of verrucae and previous treatments will be summarised. Continuous variables will be reported using descriptive statistics (eg, mean and SD), and for categorical data we will report numbers and percentages.
Primary analysis
The primary outcome is complete clearance of the index verruca at 12 weeks, which will be a dichotomous outcome (presence or absence of verruca). The proportions of participants with complete clearance of the index verruca will be compared using a χ2 test.
Secondary analysis
A logistic regression model will be used to adjust the primary analysis for three important prognostic variables (age, whether or not the verrucae have been previously treated and type of verruca). ORs and corresponding 95% CIs will be provided.
Complete clearance of all verrucae at 12 and 24 weeks as determined by a blinded assessor, participant self-reported clearance of verrucae at 12 and 24 weeks, and recurrence of verrucae at 24 weeks will be analysed in the same way as the primary outcome.
At 12 and 24 weeks, an assessment of whether the verrucae have cleared will be made by both the participant and the blinded assessor. The Cohen's κ measure of inter-rater agreement will be used to assess the agreement between the two assessments at each time point.
Poisson, or negative binomial, regression as appropriate will be used to compare the number of verrucae at 12 and 24 weeks between the two treatment groups, with adjustment for the number of verrucae at baseline.
Self-reported time to clearance of all verrucae in days from randomisation will be analysed using a Cox proportional hazards model adjusting for age, whether or not the verrucae have been previously treated and type of verruca. Participants will be right censored if they are lost to follow-up or if their verrucae have not cleared, as appropriate.
Data on side effects of treatment, pain intensity after treatment, use of painkillers, treatment details and participant satisfaction with treatment will be summarised and descriptive statistics provided.
Missing data
The amount of missing baseline data should be minimal as this will be checked by the podiatrist at the first appointment prior to the participant being randomised. We will try and minimise any missing data with respect to the primary outcome of index verruca clearance within 12 weeks. However, if we are unable to ascertain the status of any patients, then they will be treated as not having a cleared verruca in the primary analysis.
Health economics
The primary approach to economic evaluation will be a cost-effectiveness analysis of the trial intervention (needling) versus usual care (debridement). The evaluation will be carried out from the perspective of health services, over a time horizon of 12 weeks.
Costs associated with resource use will be calculated for each trial participant using data collected from the self-report questionnaires given to participants at 12 weeks. Resource use data will be collected in terms of number of visits to the podiatrist or other healthcare professionals for wart-related treatments. Costings will be applied using standardised costs where available, for instance, visits to NHS staff such as GPs or nurses will be costed according to standard NHS costs.14 Costs of delivering the intervention will be calculated from the trial and will use standardised costs where available, for example, for the podiatrists’ time.
The primary patient outcome for the economic analysis will be the primary trial outcome; complete clearance of the index verruca at 12 weeks. The incremental mean difference in costs between the two trial arms and incremental difference in patient outcome will be calculated to provide an incremental cost-effectiveness ratio (ICER) of cost per patient cured at 12 weeks. This will result in one of four scenarios:
Needling is less costly than debridement and leads to better patient outcomes;
Needling is more costly than debridement and has worse outcomes;
Needling is more costly than debridement but has better patient outcomes;
Needling is less costly than debridement and leads to worse patient outcomes.
If the results show scenario 1 or 2, then one treatment clearly dominates the other and there is a clear choice about the treatment that is cost-effective. If non-dominance occurs, as in scenarios 2 and 4, consideration must be given to the potential cost implications versus patient benefit to make a decision regarding cost-effectiveness. This will involve relating the incremental mean costs between the two trial arms to the incremental mean outcome as a ratio, the ICER.15 The ICER represents the additional cost per additional patient cured. Uncertainty regarding the cost-effectiveness analysis will be assessed using cost-effectiveness acceptability curves
Trial organisation and monitoring
Owing to the low risk of this study, site monitoring visits for this study will not be undertaken on behalf of the sponsors. The study will be run using the University of York Standard Operating Procedure Guidelines for clinical trials.
With the agreement of the sponsors, one independent steering and monitoring committee will be set up to undertake the roles traditionally undertaken by the Trial Steering Committee (TSC) and Data Monitoring and Ethics Committee (DMEC). This committee will comprise an independent chair who will be a clinician with expertise in podiatry, a HCPC registered podiatrist, the chief investigator and other study collaborators. The role of this committee will include the review of all SAEs which are thought to be treatment related and unexpected. The committee will meet at least annually or more often as appropriate. A Trial Management Group will be formed consisting of the chief investigator; trial coordinator; statistician; and the principal investigators at the site. Regular meetings will be held according to the needs of the trial.
Ethics and dissemination
Ethics
Participation in the study will be entirely voluntary. Potential participants will receive written information about the study which will include information about the potential benefits and risks of being involved in the trial. Potential participants will have the opportunity to discuss the information and the study with a podiatrist who is part of the research team. If a podiatrist feels that the potential participant is unable to give informed consent, then they would not be eligible to take part in the study. All participants will give written informed consent prior to entry into the study. The podiatrist will inform the participant if new information comes to light that may affect the participant's willingness to participate in the trial.
Dissemination
Outcomes will be disseminated through publication according to the CONSORT guidelines. This study protocol and results from the main trial will form the basis of academic papers. Results will be presented at international scientific conferences. Participants will be offered the opportunity to obtain a summary of the findings on completion of the study.
Conclusion
This will be the first appropriately statistically powered RCT to compare the clinical effectiveness of needling for the treatment of verrucae. The study results will provide valuable information about the impact of this procedure on the resolution of verrucae and therefore inform patients and healthcare practitioners of its efficacy. The secondary analysis at week 24 will provide evidence of long-term verruca clearance or reoccurrence in response to treatment.
Trial status
Recruitment and follow-up are in progress. Recruitment to the study began in March 2015 and will continue until approximately March 2016. Follow-up will be completed in September 2016 and analysis will be conducted between October and November 2016.
Acknowledgments
The research team would like to thank the independent members of the Trial Steering/Data Monitoring and Ethics Committee Professor Wesley Vernon and Belinda Longhurst for their advice, oversight of the study and reviewing of adverse event data.
Footnotes
Contributors FH, DT, MC, MH-B, CF, KB and SC wrote the protocol. All authors read and approved the final manuscript.
Funding This study is funded by the University of Salford. The University of Salford is the study sponsor and is legally responsible for the initiation and management of the study. The sponsor representative is Kay Hack.
Competing interests None declared.
Patient consent Obtained.
Ethics approval Ethical approval has been obtained from the University of Salford, Department of Health Sciences Ethical Approval Committee (HSCR15/24) and the University of York, Department of Health Sciences Research Governance Committee (HSRGC/2014/98/B).
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement All data will be published in the research manuscript at the end of the study.