Article Text

Abstract
Introduction Newly diagnosed type 2 diabetes mellitus (T2DM) in patients with coronary artery disease (CAD) more than doubles the risk of death compared with otherwise matched glucose tolerant patients. The biguanide metformin is the drug of choice in treatment of T2DM and has shown to ameliorate cardiovascular morbidity in patients with T2DM and myocardial infarction (MI). The incretin hormone, glucagon-like peptide-1 (GLP-1) improves β-cell function, insulin sensitivity and causes weight loss and has been suggested to have beneficial effects on cardiac function. The GLP-1 receptor agonist (GLP-1RA), liraglutide, is currently used for treatment of T2DM but its potential effect on cardiac function has not been investigated in detail. We hypothesised that liraglutide added to metformin backbone therapy in patients with CAD and newly diagnosed T2DM will improve β-cell function and left ventricular systolic function during dobutamine stress.
Methods and analyses 40 patients with CAD and newly diagnosed T2DM will receive the intervention liraglutide+metformin and placebo+metformin in this investigator-initiated, double blind, randomised, placebo-controlled, cross-over 12 plus 12 weeks intervention study with a 2-week washout period. The primary cardiovascular end point is changes in left ventricular ejection fraction during stress echocardiography. The primary endocrine end point is β-cell function evaluated during a frequently sampled intravenous glucose tolerance test. Secondary end points include heart rate variability, diurnal blood pressure, glucagon suppression and inflammatory response (urine, blood and adipose tissue).
Ethics and dissemination This study is approved by the Danish Medicines Agency, the Danish Dataprotection Agency and the Regional Committee on Biomedical Research Ethics of the Capital Region of Denmark. The trial will be carried out under the guidance from the GCP unit at Copenhagen University Hospital of Bispebjerg and in accordance with the ICH-GCP guidelines and the Helsinki Declaration.
Trial registrations number Clinicaltrials.gov ID: NCT01595789, EudraCT: 2011-005405-78.
- CLINICAL PHARMACOLOGY
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Strengths and limitations of this study
-
Several cardiovascular and metabolic end points will be assessed.
-
Study participants will be extensively monitored.
-
This study will contribute to a deeper understanding of data from long-term outcome studies.
-
Limited and non-representative study population.
-
No long-term outcome data will be assessed.
Introduction
The proportion of cardiovascular disease attributable to diabetes has increased over the years.1 Cardiovascular disease is the major cause of death in patients with diabetes.2 Patients with newly diagnosed type 2 diabetes mellitus (T2DM) and coronary artery disease (CAD) exhibit a twofold increased risk in mortality compared to similar glucose tolerant patients with CAD.3
The biguanide metformin ameliorates cardiovascular morbidity in patients with diabetes with acute myocardial infarction (MI)4 and total mortality in patients with obese T2DM and is therefore the first-line drug of choice in the EASD/ADA clinical guidelines.5 Furthermore, metformin improves endothelial function, insulin action and lipidemia in participants with impaired glucose homoeostasis6 and metabolic syndrome.7
The incretin hormone, glucagon-like peptide-1 (GLP-1), has several beneficial effects in diabetes, that is, it improves the β-cell function, reduces body weight, decreases circulating markers of low-grade inflammation, suppresses glucagon secretion and thereby reduces hepatic glucose production and possibly improves insulin sensitivity.8 GLP-1 has been shown to have a relaxing and vasodilatory effect on arteries in several animal studies9–11 and improves left ventricular ejection fraction (LVEF) in patients with cardiac failure,12 a finding which could not be confirmed during short-term GLP-1 treatment in a non-diabetic population.13 Moreover, GLP-1 protects the heart against ischaemic injury14 ,15 and reduces reperfusion injury in patients with ST-segment elevation MI.16 In patients with stable CAD it was suggested that a single dose of dipeptidyl peptidase 4 (DPP-4) inhibitor through increasing plasma levels of GLP-1 may improve LVEF during the dobutamine stress test.17
LVEF is regarded as a strong predictor for cardiovascular mortality in patients with CAD and diabetes. Diabetes increases the risk of left ventricular dysfunction.18 Previous studies have shown that abnormal dobutamine stress echocardiography (DSC) is associated with an increased mortality and an increased risk for cardiovascular events in patients with diabetes.19 It is not fully understood whether the physiological mechanism of GLP-1 on the heart is mediated through direct interaction on the GLP-1 receptors of the cardiovascular system or through improvement in glucose, insulin and free-fatty acids.20 However, improvement in systolic function of the left ventricle (LV) mediated through GLP-1 RA may have promising therapeutic benefits in patients with CAD and T2DM on long-term cardiovascular outcome.
Impaired β-cell function and insulin resistance are associated with metabolic syndrome, T2DM and CAD. Metformin reduces the risk of MI in patients with T2DM and the incidence of diabetes in persons at high risk by reducing insulin resistance.4 ,21 GLP-1RA exhibit pleiotropic positive effects on α- and β-cell function,22 optimise glycemic control, promote body weight loss23 and improve insulin sensitivity.24 ,25 Therefore metformin and GLP-1 RA have complementary effects on the glucose metabolism.
At present, two major randomised, controlled cardiovascular end point studies are ongoing, the LEADER26 study and the EXSCEL study,27 each including approximately 9000 patients. These studies follow patients with T2DM and increased risk of, or even evident ischaemic heart disease, for approximately 5 years and are expected to conclude in the year 2016 and 2017, respectively. Importantly, these trials are not designed to explain the mechanisms through which GLP-1RA-based treatment ameliorates T2DM or CAD. The strength of the present study is, therefore, its ability to describe a number of the pathophysiological pathways that are potentially improved following the combined therapy of metformin and liraglutide.
In conclusion, the present study examines the effects of the combined therapy of liraglutide and metformin versus metformin alone, on metabolic, inflammatory and cardiovascular parameters. The study will provide new information about the pleitropic effects of this combined treatment in patients with newly diagnosed T2DM and CAD.
Objectives
The aim is to investigate the effect of combined GLP-1RA (liraglutide) and metformin therapy on glucose metabolic and cardiovascular end points compared to metformin monotherapy in patients with CAD and newly diagnosed T2DM. It is hypothesised that therapy with GLP-1RA added to metformin in patients with CAD and T2DM will improve β-cell function and left ventricular systolic function during dobutamine stress and, in addition, improve heart rate variability (HRV) and help lower blood pressure, among other selected secondary end points. Primary and secondary end points are presented in boxes 1 and 2.
Primary end points
Cardiovascular
-
Changes in left ventricular ejection fraction (LVEF) during dobutamine stress assessed by Simpsons biplane method with use of echo contrast echocardiography17.
-
Endocrine/metabolic
-
β-cell function, that is, disposition index as measured during an insulin modified frequently sampled intravenous glucose tolerance (im-FSIGT) test according to Bergman Minimal Model approach.28
Secondary end points
Cardiovascular
-
Systolic function at rest as assessed by global strain with two-dimensional-speckle tracking.
-
Longitudinal systolic and diastolic left ventricular function at rest and during dobutamine stress using tissue velocity imaging, strain (SI), strain rate (SRI) and maximal velocity of myocardium (s′, e′, a′) and atrioventricular plane displacement.
-
Wall motion score index (WMSI).
-
Left atrial volume and surrogate markers of left ventricular filling pressure (E/e′) at rest.
-
Changes in exercise tolerance test variables: total exercise duration (s), time to limiting angina (s) and time to 1 mm ST-segment depression (s).
-
Heart rate variability (HRV) that is SDNN (standard deviation of all normal RR interval), assessed during HOLTER recording.
-
ST-depression and ectopic activity assessed during 24 h HOLTER monitoring.
-
Diurnal blood pressure.
-
Endocrine/metabolic
-
Glucagon, incretins, glucose, insulin, C-peptide response29 ,30 and β-cell glucose sensitivity during meal test
-
Insulin sensitivity (Si), acute insulin and C-peptide response29 ,30 to intravenous glucose (AIRg, ACRg), glucose clearance (Kg), glucose effectiveness (Sg) and hepatic extraction of insulin (HEXi)31 derived from an im-FSIGT (Minimal Model approach)
-
C-reactive protein, Tumour necrosis factor-α (TNF-α) and interleukin-6 (IL-6) in plasma and gene expression of IL-6 and TNF-α in subcutaneous fat
-
Non-esterified fatty acids (NEFA) during meal test32
-
NEFA during im-FSIGT by use of NEFA minimal model33
Methods and analysis
Design
A double blind, randomised, placebo-controlled, crossover, investigator-initiated, human, 12 plus 12 weeks intervention study with a 2-week wash-out period (protocol V.8, February 2013) (figure 1). Inclusion and exclusion criteria are presented in boxes 3 and 4.
Inclusion criteria
-
Stable CAD documented by one of the following;
-
Previous AMI (a minimum of 6 weeks after an acute myocardial infarct)
-
Previous coronary revascularisation
-
CAD confirmed by an abnormal coronary arteriography (CAG) or CT angiography showing stenosis >50% of any major coronary arteries.
-
-
Body mass index ≥ 25 kg/m²
-
Age ≥18 and ≤85 years
-
Type 2 diabetes mellitus (T2DM) diagnosed by one of the following:
-
HbA1c ≥6.5%
-
HbA1c <6.5% and fasting plasma glucose ≥7 mmol/L (confirmed)
-
HbA1c <6.5% and a 2 h plasma glucose value during oral glucose tolerance test (OGTT) ≥11.1 mmol/L
-
Newly diagnosed T2DM is defined as within 24 months prior to screening of the patients.
-
Exclusion criteria
-
Endocrine/metabolic
-
Type 1 diabetes mellitus defined as C-peptide <450 pM, or insulin treatment from time of diagnosis
-
Previously diagnosed diabetes for more than 24 months prior to the screening procedure for this trial, except from previous gestational diabetes
-
Simultaneous use of more than two oral antidiabetic drugs up to 3 months before screening. It is accepted that the patient continues the usual oral antidiabetic drug or is switched to metformin treatment after screening as long as the oral antidiabetic drug is paused 2 weeks before the baseline visit in the study.
-
Treatment with oral glucocorticoids, calcineurin inhibitors or other GLP-1 mimetics, which in the investigator's opinion could interfere with glucose metabolism.
-
Liver (transaminases greater than ×2 the upper normal level) or renal diseases (eGFR <60 mL/min)
-
Amylase greater than ×3 the upper reference value, or previous pancreatitis
-
Clinical myxoedema or hyperthyroid condition defined by a value of thyroid stimulating hormone (TSH) <0.1 and >10.0 mU/L
-
Anaemia, leucopaenia, or thrombocytopaenia (all <85% of lower normal limit)
-
Plasma calcium-ion ≥1.45 mmol/L
-
Plasma calcitonin ≥50 ng/L
-
Cardiovascular
-
Significant heart disease (New York Heart Association (NYHA) Functional classification. >2; ejection fraction <40% or unstable angina pectoris), or known significant heart valve disease
-
Documented atrial fibrillation or atrial flutter within 6 weeks previous to the screening. Paroxysmal atrial fibrillation is accepted if sinus rhythm is achieved at the screening.
-
Uncontrolled arterial hypertension (>180/100 mm Hg) at the time of screening.
-
Of safety/logistic type
-
Pregnancy or failure to comply with contraceptional planning within 2 years, or breastfeeding
-
Abuse of alcohol or drugs, or any other co-existing condition that would make patients unsuitable to participate in the study, as deemed by the investigators
-
Any chronic medical condition to unduly increase risk for the potential enrollee as judged by study investigators
-
Use of immunosuppressive therapy in the preceding 12 months
-
Known or suspected hypersensitivity to trial product(s) or related products.
-
Cancer (except basal cell skin cancer or squamous cell skin cancer) or any other clinically significant disorder, which in the investigator's opinion could interfere with the results of the trial
-
Inflammatory bowel disease
-
Previous bowel resection
-
Clinical signs of diabetic gastroparesis
-
Patients with personal or family history of medullary thyroid carcinoma or a personal history of multiple endocrine neoplasia type 2
-
Refusal to sign informed consent

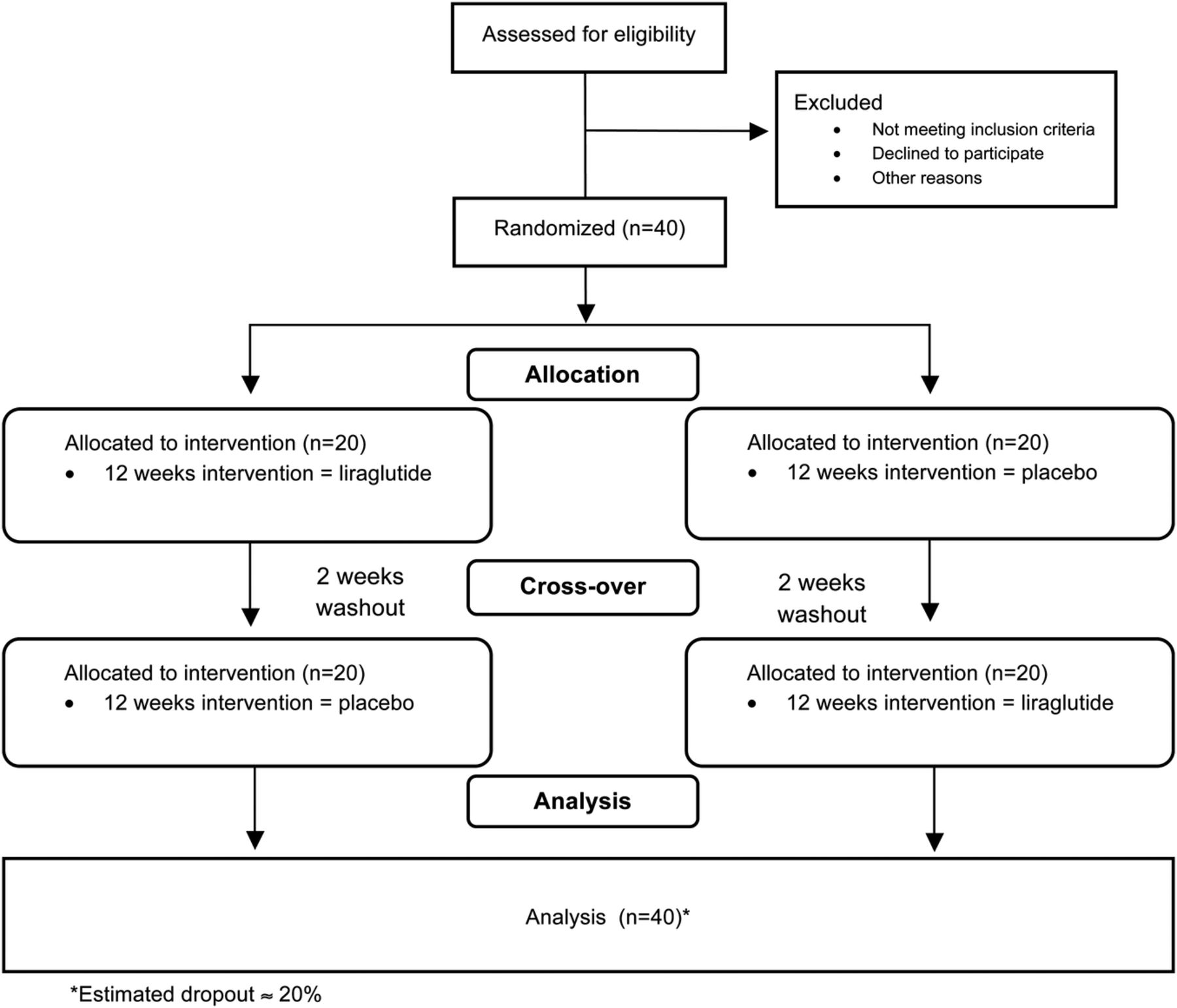{kind=link}
AddHope2 study design.
Randomisation
Forty patients meeting the inclusion criteria will be included consecutively. Patients will be identified primarily using patient files at the departments of cardiology at the Copenhagen University Hospitals, Bispebjerg and Amager, and other selected hospitals in the region. Informed consent will be obtained prior to the screening procedure. If a patient drops out of the study before completion of week 4, the patient can be replaced. Patients not meeting the inclusion criteria during the screening programme will be offered an appointment with an endocrinologist for appropriate treatment. Novo Nordisk A/S will supply liraglutide/placebo study drugs in subject boxes which are sequentially numbered with a unique code and randomised by computer in a 1:1 randomisation ratio (placebo period first or active period first). Subject boxes, which contain a total of eight pens in each period for each participant, will be given consecutively to participants. Allocation sequence will be delivered in opaque, sealed envelopes and is numbered in accordance with the unique subject box numbers. Allocation sequence will be concealed until all participants have completed the study and all the analysis which can be subject to bias has been performed.
Intervention
Liraglutide+metformin will be dosed initially as 0.6 mg+500 mg bid, after 14 days as 1.2 mg+(1000 mg+500 mg) daily and after a total of 28 days as 1.8 mg+1000 mg bid. Therapy with placebo + metformin will be titrated in an identical manner. The 2-week wash-out period has been validated for GLP-1-based therapy and fully reversed the significant effect on β-cell function.34 The dose of the study drug can be adjusted at all visits and all phone consultations if the participant experiences intolerable side effects or adverse events (abnormal blood tests related to pancreas, kidney or liver function) related to the study drug. A log of study drugs supplied to and returned from participants will be kept at the trial site.
Procedures
The study period for each patient is 26 weeks and consists of eight visits (at weeks 0, 4, 8, 12, 14, 18, 22 and 26). Wash-out will be between weeks 12 and 14. At each visit and during phone consultations in between visits the participant will be assessed for side effects, adverse events and compliance to the study drug. The outline of trial visits and examinations is shown in table 1.
Trial visits and examinations
Anthropometric data
At each visit the following measurements will be carried out: blood pressure, pulse, weight, waist and hip circumference, and an ECG will be recorded.
Subcutaneous fat biopsy
Tissue samples will be obtained using a Bergströms cannula, and will be snap frozen using liquid nitrogen and stored at −80°C.
Frequently sampled intravenous glucose tolerance
An im-FSIGT (frequently sampled intravenous glucose tolerance) will be performed using a standardised protocol according to the Bergman Minimal Model approach.28 ,35 ,36 After basal sampling, a glucose bolus (0.3 g/kg) is injected intravenously over 60 s. An infusion of 0.02 IU/kg insulin (Actrapid, Novo Nordisk, Bagsvaerd, Denmark or Humulin Regular, Eli Lilly, Indianapolis, Indiana, USA) is administered intravenously over 30 s at 20 min after the glucose bolus. Blood samples are taken for measurement of insulin and non-esterified fatty acid (NEFA) every 1 to 8 min, and every other minute to 30 min, every 10–100 min and at 150 and 180 min. No samples are taken at 20 min, when insulin is injected. A total of 27 blood samples will be obtained for measurement of insulin and NEFA over 180 min. C-peptide is measured only until administration of insulin. Bedside glucose measurement will be carried out at every blood sample.
Meal test
A 2 h meal test will be performed after the FSIGT. A standard meal of 550 kcal (46% carbohydrate; 28% protein; 26% fat) will be consumed within 15 min. Blood samples taken at −10, −5, −1, 30, 60, 90 and 120 min relative to commencing the meal34 will be analysed for glucose, insulin, C-peptide, NEFA, GLP-1, gastric inhibitory polypeptide, glucagon and triglyceride.
Echocardiography
Two-dimensional echocardiography (Vivid E9, GE Vingmed Ultrasound, Horten, Norway) will be performed at rest using a M5S transducer. Parasternal long-axis and short-axis views, apical four-chamber, two-chamber and long-axis views and apical view of the right ventricle will be captured with patient supine in left lateral position. Recordings with focus on left atrium (LA) will be performed in apical four-chamber and two-chamber views. Pulsed wave Doppler (PW-Doppler) velocities of mitral inflow will be recorded with sample volume placed at the tip of the mitral leaflets. Simultaneous PW-Doppler velocities of the aortic and mitral flows will be recorded for assessment of isovolumic relaxation time. Supplementary recordings will be optimised for speckle tracking in apical four-chamber, two-chamber and long axis views of the LV with a frame rate of about 70±10/s. Colour tissue Doppler imaging (c-TDI) of both the right and left ventricle will be captured with a frame rate of 200±20/s. All recordings will be made in midexpiration to minimise beat-to-beat variability.
Dobutamine stress echocardiography with echo contrast
DSE will be performed with the patient supine in the left lateral position. Under continuous ECG monitoring, dobutamine will be administered using an infusion pump into a peripheral vein at an incremental regimen of exact 10 µg/kg current body weight per minute increased every 3–5 min up to a maximum of 40 µg/kg/min. Patients will be asked to omit their β-blockers for 36–48 h before each scan. Atropine 0.3 mg (up to a maximum of 2 mg) will be administered if necessary to achieve target heart rate (HR). 2D echocardiography will be performed at rest (baseline), at 10–20% increase in HR above baseline (low dose), at 85% of age-predicted maximum HR (220—age in years) (peak dose) and after terminating the dobutamine infusion when the HR has declined to low dose level (recovery). Systolic and diastolic blood pressure will be assessed at baseline and at the end of each stage. Three cardiac cycles at rest, and five cardiac cycles during stress will be captured. Ultrasound contrast agent SonoVue (Bracco International, Amsterdam, Netherlands) or Optison (GE Healthcare, Princeton, New Jersey, USA) will be administered into the right antecubital vein during all stages of the dobutamine infusion. Contrast-specific imaging modalities will be employed involving adjustment of the power output to intermediate mechanical index (MIX) harmonic imaging (MIX 0.3, gain 2, compress 20%, reject 0, dynamic range 9). Between each step of dobutamine stress, a high MIX will be used to eliminate microbubbles before the next stress level. Apical four-chamber and two-chamber views will also be captured with c-TDI without contrast. The image sector will be kept as narrow as possible in order to achieve a c-TDI frame rate between 180 and 200 frames/s. Modified criteria will be used for terminating dobutamine infusion.37 ,38 Absolute criteria are: achievement of target HR ((220−age)×0.85 bpm); new or worsening wall motion/thickening abnormalities in at least two adjacent LV segments; and severe chest pain. Relative criteria are: non-tolerable symptoms or increase in systolic blood pressure to >240 mm Hg or decrease to <100 mm Hg; blood pressure decrease > 40 mm Hg below baseline systolic blood pressure or >40 mm Hg decrease from previous level; frequent arrhythmias such as supraventricular tachycardia, atrial fibrillation or frequent polymorphic ventricular beats; and ischaemic ECG changes.
Exercise tolerance test
A standard exercise tolerance test on a cycloergometer with continuous 12-lead ECG monitoring will be performed. The workload will be increased by 25 or 50 W as convenient in intervals of 2 min. Participants will be encouraged to continue until maximal exhaustion.
Diurnal blood pressure
Diurnal blood pressure will be recorded (Model TM-2430, A&D Co Ltd, Japan) with 30 min intervals between 6:00 to 22:00 and 60-min intervals during night time.
Holter monitoring
Holter monitoring for up to 48 h will be performed (Rozinn RZ 153+12, Rozinn Electronics).
Analysis
Blood and tissue samples
Subcutaneous fat biopsy and blood samples drawn during FSIGT and meal test will be aliquoted and stored at –80°C for later analysis. Bedside glucose measurements will be carried out on Accu-Chek Inform II (Roche Diagnostics GmbH, Mannheim, Germany). At each visit patient samples of full blood, serum, plasma and urine will be obtained and stored at −80°C for future additional analysis.
Analysis of the exercise tolerance test
The following variables will be assessed: total exercise duration (s), maximum exercise HR and blood pressure, maximum rate pressure product (heart rate×blood pressure), time to limiting angina (s) and time to 1 mm ST-segment depression (s). Total metabolic equivalents (METS) achieved, ‘Heart-rate recovery’ 1 min after termination of exercise.
Analysis of the diurnal blood pressure
Mean 24 h blood pressure, and mean daytime and night-time blood pressure will be assessed.
Analysis of the Holter monitoring data
Analysis will be performed using Holter for Windows+ (ScottCare Corporation, Cleveland, Ohio, USA). The following variables will be analysed: HR (resting, daytime and night-time) and HRV; and the following variables from time domain and frequency domain measures of HRV are preselected: MEANNN and SDNN (mean value and SD of all normal RR intervals); MEANN will be representing the average 24 h HR, that is, 60.000/MEANNN = average 24 h HR. SDNN-index (mean of the standard deviations of all NN intervals for all 5 min segments of the entire recording), RMSSD (the square root of the mean of the sum of the squares of differences between adjacent NN intervals), SDANN (the standard deviation of the average of normal-to-normal intervals for each 5 min period) and the percentage of adjacent cycles that are >50 ms apart (pNN50). Furthermore, very low frequency (VLF power: <0.04 Hz,) low frequency (LF power: 0.04–0.15 Hz), high frequency (HF power: 0.15.0.4 Hz), LF/HF and total power will be assessed.
Analysis of the echocardiographic images
A reviewer blinded to the treatment strategy will analyse the echocardiographic images off-line (GE EchoPAC V. 112). LV internal dimensions, interventricular septal and posterior wall thickness will be measured in 2D at the tip of the mitral valve.39 Using the biplane method of Simpson in two-chamber and four-chamber views the LV volume, LVEF and cardiac output will be calculated from the contrast enhanced images. LA volume will be calculated using the biplane area length method.39 LV mass will be measured using the area–length formula.39 Regional LV wall motion will be assessed using a 16-segment model that comprises basal and septal levels of six regional walls (anterior, lateral, posterior, inferior, septal, anterior septal) and apical level of four regional walls (anterior, lateral, inferior, septal).39 Early diastolic filling velocity (E), late atrial filling velocity (A), E-wave deceleration time (EDT) and E/A ratio will be assessed from the PW-Doppler velocity recording of the mitral inflow. Isovolumic relaxation time will be measured from the onset of the aortic valve closure signal to the onset of mitral inflow signal from the simultaneous PW-Doppler recording of the aortic and mitral flows. Peak early diastolic annular velocities (e′), late diastolic velocities (a′) and peak systolic annular velocities (s′) will be measured from c-TDI images with sample area placed in the septal, lateral, anterior and inferior parts of the mitral annulus and from the septal and lateral parts of the tricuspid annulus. Measurements will be averaged from three beats. Measurement of tricuspid annular plane systolic excursion will be obtained from an M-mode image through the junction of tricuspid annulus and free wall of right ventricle. Systolic mitral annular excursion will be measured in reconstructed longitudinal M-mode and from tissue Doppler by integration of the c-TDI velocity curves. The measurements will be obtained from the septal, lateral, anterior and inferior walls and averaged from three beats. Global strain and segmental values of peak longitudinal strain and peak longitudinal strain rate will be assessed from apical four-chamber and two-chamber, and long-axis views optimised for speckle tracking with a frame rate about 70±10/s. Basal strain and strain rate will be assessed from c-TDI images in the dobutamine stress protocol with sample volumes placed in the basal part of the septal, lateral, anterior and inferior myocardial walls. Sample volume will be adjusted to track the region of interest over three consecutive beats using anchor points in the off-line analysis. Wall motion score will be assessed using a 16-segment model of the left ventricle.40 Each visualised segment will be graded according to the following score: 1=normal; 2=hypokinetic; 3=akinetic; 4=dyskinetic; and 5=aneurysmal. WMSI will be calculated by dividing the sum of scores by the number of segments visualised.
Statistical analysis
Primary reporting of end points will be based on the basic estimator notion41 ,42 controlling for period effects and assuming no period by treatment interactions, including carry-over effects. Patient effects will be modelled using the random effects. Liraglutide is, however, expected to induce significant weight loss, which could confound the effect of the backbone therapy in the group that is treated with liraglutide in the first period. We recognise that the use of baseline measurements before each treatment period does not secure the analyses against bias from such a carry-over effect. Therefore secondary analyses utilising modelling of the carry-over effect is also planned as sensitivity analyses. Normalising transformations will be applied as appropriate. Analyses will include all data on all patients, regardless of completion status, that is, patients dropping out will contribute with whatever data are available.
Power calculation
Given a similar positive effect of liraglutide on β-cell function in CAD patients with newly diagnosed T2DM, as was shown by vildagliptin (a DPP-4 inhibitor) for patients without CAD with impaired glucose homoeostasis (+69% in disposition index),34 n=22 patients in paired analyses provide a statistical power of 80% at p<5% . Given a similar effect of liraglutide treatment during 12 weeks as compared to a single dose of sitagliptin on stress induced LVEF in patients with CAD (+8.7%), n=14 patients in paired analyses would provide a statistical power of 90% at p<5%.17 For one of the secondary end points, HRV: given a similar positive effect of liraglutide therapy on HRV (SDNN) (+29%) as was shown by a very low calorie dietary intervention during 6 weeks in insulin resistant non-diabetic obese participants, n=16 patients in paired analyses would provide a statistical power of 90% at p<5%.43 The improvement during liraglutide therapy on metabolic features, which probably drives the improvement in autonomic nervous function resulting in improved HRV,43 is comparable to what has been achieved during very low calorie dietary intervention on HRV.44 The SDs across the study sample and the intra-individual variances obtained in these prior studies on the end points: β-cell function, stress-induced LVEF and HRV, respectively,9 ,34 ,43 would most likely be quite similar in the present study as compared to these prior studies. Thus, if n=40 patients with CAD and T2DM are randomised in ratio 1:1 to metformin versus metformin in combination with a GLP-1 analogue in a cross-over design and a drop-out rate during 26 weeks of approximately 20% is estimated, that would provide n=32 patients to be evaluated in paired analysis. This will result in a statistical power ≥90% for the primary end points at significance level p<0.05. Effect of intervention is calculated by ANCOVA analysis using baseline values as co-variates.
Ethics and dissemination
The study will be carried out according to the ICH-GCP (International Conference on Harmonisation—Good Clinical Practice) standards and will be monitored by the GCP-unit at Copenhagen University Hospital of Bispebjerg. The AddHope2 trial will provide the basis for two PhD fellowships, one covering metabolic and one covering cardiovascular end points. Positive and negative study results will be published in peer-reviewed international medical journals. None of the funders have any influence on the decision to publish the work or on the content of the work to be published.
Timeframe
Screening, randomisation and baseline examinations are performed from May 2012 to April 2014. Follow-up (12/14/26 weeks examinations) are performed from June 2012 to October 2014. Data acquisition and analyses of data are performed from November 2014 to August 2015.
Safety considerations
Relevant concomitant care and treatment will be given if the participant experiences any adverse events or side effects during the trial related or not related to the study drug or trial procedures, for example, severe hypoglycaemia or hyperglycaemia, severe hypotension or hypertension or severe arrhythmia. Participants with a verified fasting blood glucose >13.3 mmol/L will be withdrawn from the study and offered supplementary antidiabetic treatment. Participants who experience unstable angina pectoris will be withdrawn and offered treatment according to clinical guidelines. A participant will be withdrawn from the study if continuous (lasting more than 8 weeks) observations of the following are seen: reduction in glomerular filtration rate <45 mL/min, elevation of transaminases >3 times the upper reference value, elevation in p-amylase >3 times the upper reference value, reduction in haematological parameters below 75% of the lower reference value or paroxysmal atrial fibrillation.
Acknowledgments
The AddHope2 trial would like to thank Actavis A/S (Gentofte, Denmark) for the supply of metformin tablets. The Tectra unit of Capital Region of Denmark has approved and helped in issuing the research contract with Novo Nordisk A/S. The GCP unit of Capital Region of Denmark has supervised and will continue to supervise the study in accordance with the GCP guidelines.
References
Footnotes
CA and PK contributed equally.
-
Collaborators Christian Lange, MD, Department of Internal Medicine, Amager Hospital, University of Copenhagen, Denmark; Jens Juul Holst, MD, DMSc, Department of Biomedical Sciences, The Panum Institute, University of Copenhagen, Denmark; Jens Mølvig, MD, DMSc, Department of Internal Medicine, Amager Hospital, University of Copenhagen, Denmark; Carolyn Deacon, PhD, DMSc, Department of Biomedical Sciences, The Panum Institute, University of Copenhagen, Denmark; Mogens Fenger, MD, DMSc, Department of Clinical Chemistry, Hvidovre Hospital, University of Copenhagen, Denmark; Ove Andersen, MD, PhD, Clinical Research Centre, Hvidovre Hospital, University of Copenhagen, Denmark; Raymond Boston, PhD, Department of Clinical Studies, NBC, School of Veterinary Medicine, University of Pennsylvania, PA, USA.
-
Contributors CA and PK are equal contributors to this manuscript. SBH and AS have contributed to conception and design of the trial and will contribute to the analysis and interpretation of data. SBH is the sponsor. AS is the principal investigator. CA, PK, MSK, OW and OPK are co-investigators at the Department of Cardiology, University Hospital of Bispebjerg and will contribute to the acquisition, analysis and interpretation of data. SM and SL will contribute to the analysis and interpretation of data. All authors have been involved in drafting and revising the manuscript, and have given final approval of this manuscript.
-
Funding This study is supported by a grant for investigator-initiated studies from Novo Nordisk A/S. Additional funding is provided by The AP Moller Foundation, The Bispebjerg Hospital Research Foundation, The Danish Heart Foundation, Department of Internal Medicine at Amager Hospital and Clinical Research Centre Hvidovre Hospital.
-
Competing interests SBH has received funding of educational and research tasks from Novo Nordisk, Abbott, Eli Lilly, Pfizer, Boehringer-Ingelheim, Bristol-Meyers Squibb and Merck, Sharp & Dome. OW has received funding of educational and research tasks from Resmed and Servier. SM has during the past 3 years participated in advisory boards for Novartis Pharma, Novo Nordisk, Merck, Sharp & Dome, Sanofi-Aventis, AstraZeneca, Johnson & Johnson, Roche, Mankind, Boehringer-Ingelheim, Zeeland, Eli Lilly, Intarcia Therapeutics and has received honorary for lectures from Novo Nordisk, Merck, Sharp & Dome, AstraZeneca, Johnson and Johnson, Roche, Shering-Ploug, Sanofi-Aventis, Novartis Pharma, Eli Lilly and Bristol-Meyers Squibb.
-
Ethics approval The study has been approved by the Regional Committee on Biomedical Research Ethics of the Capital Region of Denmark and publications will be in accordance with Danish law on processing of personal data and will follow the commitments of Vancouver and the CONSORT Statement.45
-
Provenance and peer review Not commissioned; peer reviewed for ethical and funding approval prior to submission.
-
Data sharing statement No additional data available.