Article Text

Abstract
Introduction Many patients with ulcerative colitis (UC) present with acute exacerbations needing hospital admission. Treatment includes intravenous steroids but up to 40% of patients do not respond and require emergency colectomy. Mortality following emergency colectomy has fallen, but 10% of patients still die within 3 months of surgery. Infliximab and ciclosporin, both immunosuppressive drugs, offer hope for treating steroid-resistant UC as there is evidence of their short-term effectiveness. As there is little long-term evidence, this pragmatic randomised trial, known as Comparison Of iNfliximab and ciclosporin in STeroid Resistant Ulcerative Colitis: a Trial (CONSTRUCT), aims to compare the clinical and cost-effectiveness of infliximab and ciclosporin for steroid-resistant UC.
Methods and analysis Between May 2010 and February 2013, 52 UK centres recruited 270 patients admitted with acute severe UC who failed to respond to intravenous steroids but did not need surgery. We allocated them at random in equal proportions between infliximab and ciclosporin.The primary clinical outcome measure is quality-adjusted survival, that is survival weighted by Crohn's and Colitis Questionnaire (CCQ) participants’ scores, analysed by Cox regression. Secondary outcome measures include: the CCQ—an extension of the validated but community-focused UK Inflammatory Bowel Disease Questionnaire (IBDQ) to include patients with acute severe colitis and stoma; two general quality of life measures—EQ-5D and SF-12; mortality; survival weighted by EQ-5D; emergency and planned colectomies; readmissions; incidence of adverse events including malignancies, serious infections and renal disorders; disease activity; National Health Service (NHS) costs and patient-borne costs. Interviews investigate participants’ views on therapies for acute severe UC and healthcare professionals’ views on the two drugs and their administration.
Ethics and dissemination The Research Ethics Committee for Wales has given ethical approval (Ref. 08/MRE09/42); each participating Trust or Health Board has given NHS Reseach & Development approval. We plan to present trial findings at international and national conferences and publish in high-impact peer-reviewed journals.
Trial registration number ISRCTN: 22663589; EudraCT number: 2008-001968-36
- Ulcerative colitis
- Acute severe
- Steroid refractory
- Ciclosporin
- Infliximab
- Randomised controlled trial
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Strengths and limitations of this study
-
A multicentre, UK-wide, pragmatic randomised trial of 270 participants recruited at 52 sites.
-
Use of mixed methods to compare clinical and cost-effectiveness and assess professional and patient views.
-
Formal follow-up using designed data collection for up to 3.5 years after randomisation and routine data for 10 years.
-
Study limited to the UK.
-
Rigorous blinding limited to main study team including analysts.
Introduction
This paper summarises the current approved protocol in use at 68 participating centres across the UK.
Background
Ulcerative colitis (UC) is a chronic debilitating disease that affects about 150 000 people in the UK.1 ,2 Many patients with UC present with acute severe colitis requiring inpatient admission. Treatment includes intravenous steroids but about 40% of patients are resistant to steroids. In the past when no other treatments were available, emergency colectomy was the only option. Although mortality following emergency colectomy has fallen over time, 10% of patients die within 3 months of surgery.3
Infliximab and ciclosporin are immunosuppressive drugs that offer hope for the treatment of steroid-resistant UC. There is evidence that they are both effective in the short term,4 particularly among patients who respond partially to steroid treatment.
Several studies support the use of infliximab in patients with moderate or severe UC,4–7 especially steroid-resistant patients who do not tolerate ciclosporin.6 A systematic review and meta-analysis of 34 infliximab studies found an average short-term response and remission of 68% and 40%, respectively, and an average long-term response and remission of 53% and 39%, respectively.7 Two large randomised controlled trials (RCTs) also found highly significant improvements in total Inflammatory Bowel Disease Questionnaire (IBDQ) scores and Short Form-36 physical and mental component scores for infliximab patients at 8 weeks when compared with placebo.8 Current UK National Institute for Health and Care Excellence (NICE) guidelines allow the use of infliximab only when ciclosporin is contraindicated or as part of a research study.9
Several studies support the use of ciclosporin as a safe and effective treatment for steroid-resistant UC,10–12 although it has side effects including dose-related toxicity11 ,13 ,14 and long-term failure.11–13 ,15 A systematic review and meta-analysis of 16 ciclosporin studies reported a mean short-term response rate of 73% but poor long-term response rates11; one study reported that 65% of patients relapsed after 1 year, 90% relapsed after 3 years13 and 58% needed colectomy after 7 years.15 Another review of 32 studies reported a 51% short-term success rate.16 However, the relevant Cochrane review concluded that there was limited evidence that ciclosporin was more effective than standard treatment for severe UC and that long-term benefits were unclear.17 It also advocated research into the long-term effects of ciclosporin on quality of life and cost-effectiveness.
A recent meta-analysis of 361 patients in six retrospective cohort studies concluded that infliximab and ciclosporin are comparable when used as rescue therapy in acute severe steroid-resistant UC. However, the outcome measures were limited to colectomy rates, adverse drug reactions and postoperative complications over 12 months.18 A recent meta-analysis of 38 patients with steroid-resistant UC showed that the 20 on ciclosporin had a higher colectomy rate, more steroid dependency and more adverse events (AEs) than the 18 on infliximab.19
Against this background of studies that compared these two drugs only indirectly, la Groupe d'Etude Thérapeutique des Affections Inflammatoires Digestives (GETAID) recently reported on the trial ‘CYcloSporine versus InFliximab (CYSIF)’,20 the first head-to-head comparison of these two drugs. CYSIF found no significant differences in ‘treatment failure’ within 98 days, defined as any of: (1) no clinical response after 7 days; (2) no remission without steroids after 98 days; (3) relapse between 7 and 98 days; (4) serious AE leading to treatment interruption; (5) colectomy or (6) death. However, CYSIF recruited only 110 patients, followed them for only 98 days, reported no data on quality of life, and collected no data on costs.
In summary, infliximab and ciclosporin are often effective in the short term, but there is little long-term evidence, especially about comparative clinical and cost-effectiveness. The Evidence Review Group Report commissioned by NICE21 concluded: ‘The results consistently indicate that the move from standard care to ciclosporin is highly cost-effective. Thus the policy question is clear: should we make a subsequent move from ciclosporin to infliximab.’ Hence we have designed Comparison Of iNfliximab and ciclosporin in STeroid Resistant Ulcerative Colitis: a Trial (CONSTRUCT) to achieve a rigorous and comprehensive comparison of these costly therapies.
In response to this debate, the CONSTRUCT team has recently extended the measurement of quality of life and costs in four ways:
-
By extending data collection for all trial participants, whenever recruited, until early 2014;
-
By adding questionnaires at 18, 30 and 36 months;
-
By adding four questionnaires following colectomy and any ensuing corrective surgery;
-
By planning to use survival analysis, statistical missing value imputation and economic modelling to impute quality of life and costs for all participants who generate data after randomisation.
Aim and objectives
The aim of this trial is to compare the clinical and cost-effectiveness of infliximab and ciclosporin for patients with steroid-resistant UC over a period of up to 3.5 years.
Specific objectives are to:
-
Compare health-related quality of life between the two treatments (infliximab and ciclosporin).
-
Compare mortality, disease activity and morbidity between treatments.
-
Compare (emergency) colectomy rates between treatments.
-
Compare cost-effectiveness of the two treatments in terms of cost per quality-adjusted life-year.
-
Extend this comparison by modelling lifetime costs and effects.
-
Investigate the views of patients about these treatments.
-
Investigate the views of healthcare professionals about the drugs, and their administration and ease of handling.
Methods
Trial design
CONSTRUCT is a two-arm, pragmatic randomised trial (figure 1). Participating patients and their doctors knew which of the two drugs they were receiving. As the two modes of administration are different, any other approach would have been difficult. Fortunately this pragmatic approach enables us to compare the clinical and cost-effectiveness of the drugs in normal clinical practice, whereas the more explanatory double-blind placebo controlled trial would have compared the efficacy of the drugs under stricter, less realistic, conditions.22
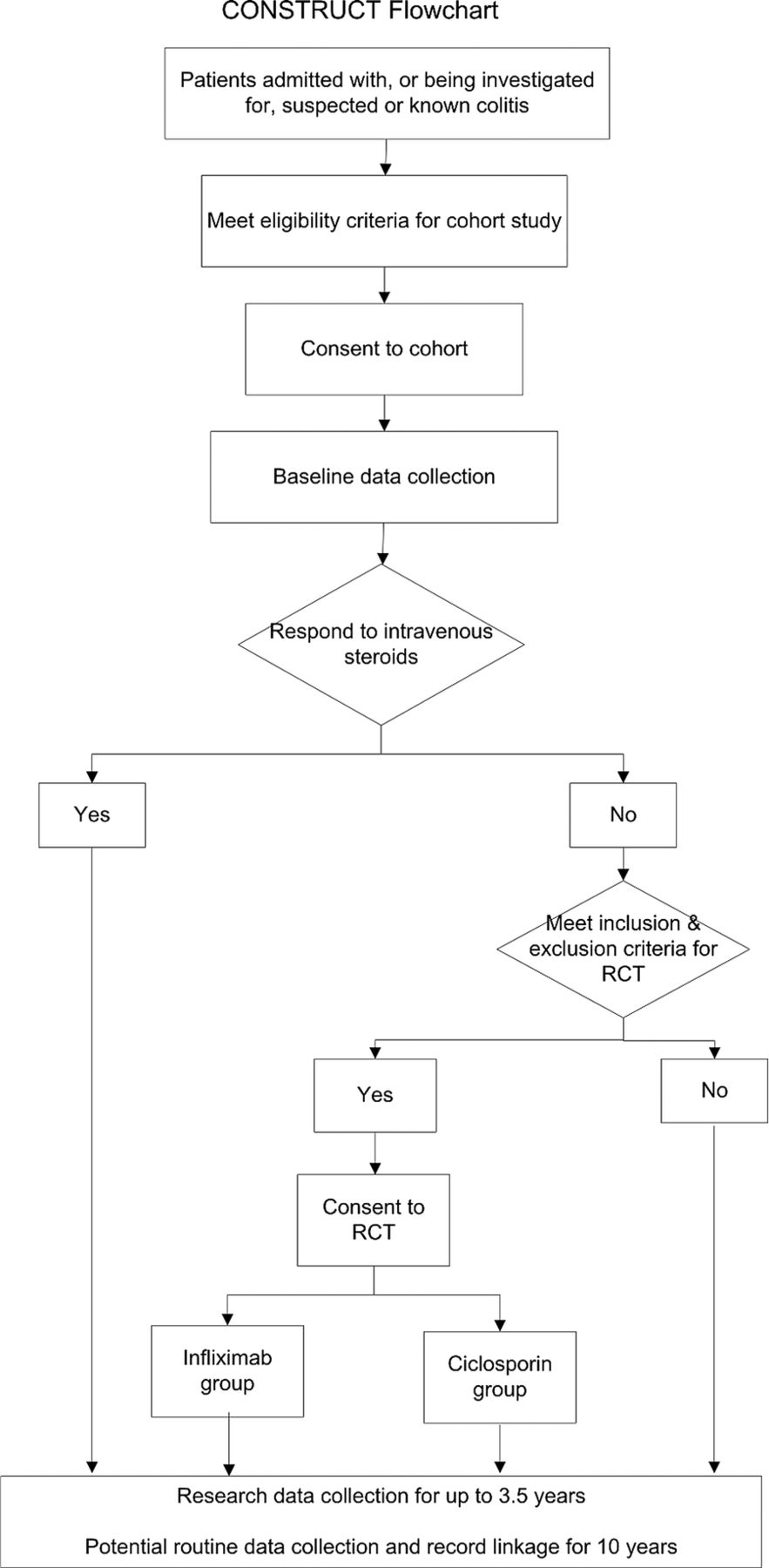
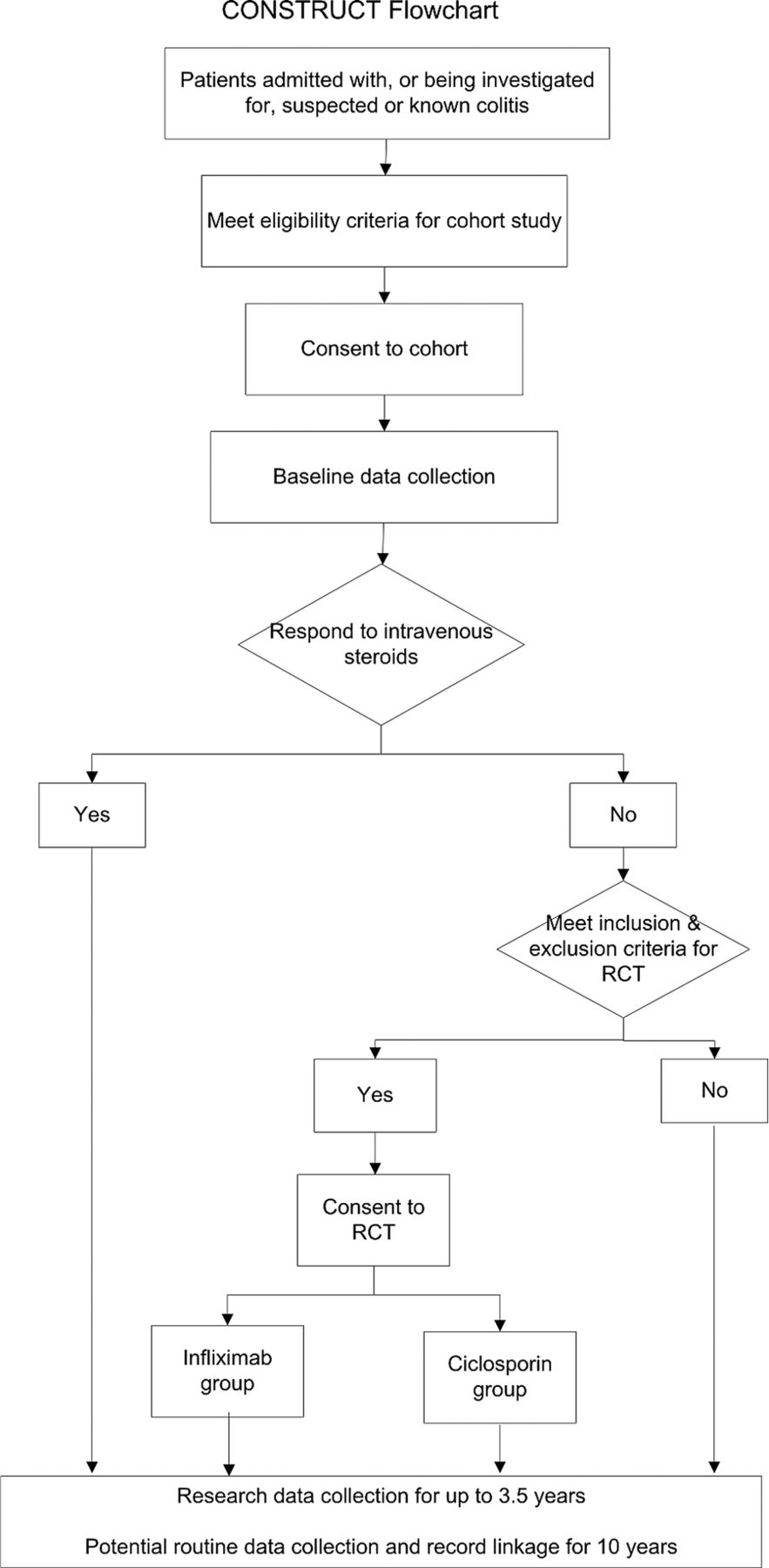{kind=link}
Comparison Of iNfliximab and ciclosporin in STeroid Resistant Ulcerative Colitis: a Trial (CONSTRUCT) flowchart.
To this design we added a comprehensive cohort to help recruit patients into a complex trial. Following explanation we invited inpatients with known or suspected colitis to join the cohort as soon as possible after admission. We told them that if they had UC and did not respond to intravenous steroids, they might need other drug treatment; and, if they were suitable, we would invite them to have further treatment as part of a clinical trial. To increase the chance of recruiting them, and ensure the quality of their data, we collected their baseline data as soon as possible after they had given consent. Although our primary aim in recruiting this cohort was to enhance recruitment to the trial, it has potential to enhance future study of UC, notably through anonymised linkage to routine data.23
From May 2010 to the end of February 2013 we recruited from the cohort to the trial those participants diagnosed with UC (assessed by Truelove and Witts criteria,24 a Mayo score of at least 2 on endoscopy, or clinical judgement) who failed to respond to a course of up to about 5 days of intravenous steroids but did not need immediate surgery. After the clinical team had confirmed steroid resistance, and full oral and written explanation, we invited participants who fulfilled the trial inclusion criteria to consent to randomisation to either infliximab or ciclosporin. A placebo control group would have been unethical, as these patients are severely ill and need treatment, as the NICE Evidence Review Group recognised.21
Sample size and power
The current design of CONSTRUCT requires a combination of survival analysis and statistical imputation to get full value from the resulting dataset. As these techniques are difficult to incorporate into power calculations, we present a simpler calculation based on t tests of mean Crohn's and Colitis Questionnaire (CCQ) scores at 12 months. For CONSTRUCT to detect an effect size of 0.35 in these scores (ie, a difference between infliximab and ciclosporin groups of at least 0.35 of the population SE) with 80% power when using a 5% significance level requires that we analyse at least 250 of the 270 participants recruited to the trial. Although more than 10% of CONSTRUCT participants are likely to drop out over the follow-up period of at least 12 months, all analyses will exploit the techniques of statistical imputation used successfully by the Cancer of the Oesophagous or Gastricus: New Assessment of the Technology of Endosonography (COGNATE)25 and Folate Augmentation of Treatment—Evaluation of Depression (FolATED)26 trials to maintain the effective sample size at 250. Our initial, more conservative power calculation had proposed: to recruit 480 participants; not to impute missing data statistically; therefore to allow for 25% loss to follow-up; and thus to yield 80% power to detect a slightly smaller effect size of 0.30 in CCQ scores. We amended our target to 270 participants once the difficulty of identifying patients with acute steroid-resistant UC became clear early in recruitment.
Routine data
Designed research data collection will continue until April 2014. We shall supplement the designed data collected from cohort participants by operational data from their electronic records, routine data on National Health Service (NHS) resource use from Hospital Episode Statistics (HES) and mortality data from the Office for National Statistics (ONS).
We plan to continue follow-up for up to 10 years through record linkage of routine mortality, inpatient and primary care data using the existing facilities of the Medical Research Council (MRC)-funded Centre for Improving Population Health through E-health Research (CIPHER) in the College of Medicine of Swansea University. We hope to monitor all participants’ long-term outcomes, including mortality, emergency colectomy, elective colectomy and major morbidity including hospitalisation and surgery, and thus most of their NHS costs. Thus we aim to achieve both long-term follow-up for trial participants and to create a larger electronic comprehensive cohort of patients with inflammatory bowel disease (IBD).
Trial management
The management structure includes: Trial Steering Committee (TSC) which provides oversight of the trial and meets at six monthly intervals, Data Monitoring and Ethics Committee (DMEC) which monitors trial data in accordance with an agreed charter and reports to the TSC, and Trial Management Group (TMG) comprising academics, health professionals, researchers and representatives of service users and the Swansea Clinical Trials Unit—the West Wales Organisation for Rigorous Trials in Health (WWORTH). The TMG undertakes general management of the trial, meets every month and reports to the TSC. In accordance with the relevant WWORTH standard operating procedure,27 we include service users as active contributors at all stages of this study. They attend the TSC, DMEC and TMG where they contribute to the research process and provide valuable insights into UC.
Setting and site selection
We invited consultant gastroenterologists to express interest in taking part in the study via the British Society of Gastroenterology by completing a feasibility questionnaire. We considered sites which had treated four or more patients with steroid-resistant UC in the previous 12 months to be eligible and invited them to seek local approval. As a result we are undertaking the study in 68 NHS Health Boards or Trusts, including large teaching and district general hospitals in England, Scotland and Wales.
Informed consent
Patients who were eligible for the cohort received Cohort Participant Information Leaflets and oral explanation about the cohort from consultant gastroenterologists or research professionals (usually research nurses); they gave written consent by signing and dating a Cohort Consent Form. Cohort participants who became eligible for the trial received Trial Participant Information Leaflets and oral explanation about the trial; they gave written consent by signing and dating a Trial Consent Form. For cohort and trial, those taking consent countersigned and dated the form to confirm that the participant had fully understood the nature of the study and had had opportunity to ask questions; they also put a copy of that consent in the medical record, and gave another copy to the participant.
Research professionals could take consent to the cohort if authorised to do so on the Site Delegation Log following appropriate training including Good Clinical Practice (GCP). Although they could also explain the trial to cohort participants, responsibility for countersigning lay with the site principal investigator (PI) or another doctor with delegated authority on the Site Delegation Log.
Withdrawal
The process of consenting participants stressed that they could withdraw from cohort or trial whenever they wished without giving a reason and without affecting their care in any way. We documented reasons given where available. We encouraged participants who withdrew from completing questionnaires to continue other types of follow-up. We encouraged site staff to trace participants lost to follow-up and document the reasons for their loss when possible.
Clinical judgements that participants had not responded to treatment with infliximab or ciclosporin usually led to surgical intervention; however, those participants remained in the trial, especially for analysis by treatment allocated, unless they chose to withdraw. Between randomisation and the end of the trial, there were many decisions made by clinicians to discontinue or change allocated treatments; again this did not constitute withdrawal from this pragmatic trial.
Trial inclusion criteria
Patients admitted as emergency admissions with severe colitis who fail to respond to up to about 5 days of intravenous hydrocortisone therapy, who also have either:
-
Histological diagnosis of UC in this episode;
-
Histological diagnosis of indeterminate colitis in this episode, where clinical judgement (based on macroscopic appearance, disease distribution or a history) suggests a diagnosis of UC rather than Crohn's disease;
-
Symptoms typical of UC while histology awaited;
-
History of UC (previously confirmed histologically).
We withdrew from treatment participants with B, C or D who were later found not to have UC but kept them in the trial and followed them up in the usual way.
Trial exclusion criteria
-
Age under 18 years on admission
-
Pre-randomisation histological diagnosis inconsistent with UC (cf inclusion criterion ‘B’)
-
Enteric infection confirmed before randomisation by stool microscopy, culture or histology (includes Campylobacter, Clostridium difficile, Cytomegalovirus, Salmonella and Shigella)
-
Vulnerable patient
-
Unable to consent
-
Positive pregnancy test or currently lactating
-
Woman of child-bearing potential but not prepared to use contraception during, and for 6 months after, treatment with infliximab in accordance with the Summary of Product Characteristics
-
Current malignancy, excluding basal cell carcinoma
-
Serious co-morbidity, including:
-
Immunodeficiency
-
Myocardial infarction (within last month)
-
Moderate or severe heart failure (New York Heart Association (NYHA) class III or IV)
-
Acute stroke (within last month)
-
Respiratory failure
-
Renal failure
-
Hepatic failure
-
Active or suspected active tuberculosis
-
Other severe infections such as sepsis, abscesses and opportunistic infections
-
History of hypersensitivity to infliximab, ciclosporin or polyethoxylated oils (Sandimmun Concentrate for intravenous infusion)
-
-
Concomitant use of tacrolimus or rosuvastatin
-
English not good in absence of local translation services
-
Clinical need to undergo emergency colectomy without further medical treatment
-
Currently taking part in another clinical trial
-
Treatment with either infliximab or ciclosporin in the 3 months before admission
-
Contraindication(s) to treatment with infliximab or ciclosporin
Randomisation
We allocated all patients who completed baseline observations, met the inclusion criteria but none of the exclusion criteria, and gave informed consent—at random between infliximab or ciclosporin. We used a web-based password-protected site with a dynamic algorithm to protect against subversion while ensuring that each arm of the RCT was balanced by centre.28
For validation, the website requested additional information.
-
Participant's study number, and month and year of birth.
-
Name of the person who was requesting randomisation (limited to those trained and authorised).
-
Has consent been given?
-
Does the patient meet the inclusion criteria?
-
Does the patient have none of the exclusion criteria?
-
Has the baseline questionnaire been completed?
If the responses to the last four questions were ‘Yes’, the patient was randomised.
The research staff requesting randomisation received the name of the drug to be allocated to the participant and immediate confirmation of the study number and drug by email. The outcomes of randomisation are recorded on the randomisation database, in the trial master file at Swansea University, at the study site in participants’ records, and in the remotely hosted data repository.
The drugs were held in hospital pharmacies at trial sites. When a participant was randomised, the research staff faxed the relevant pharmacy a copy of the confirmation of participant study number and drug. The drug was labelled with the EudraCT number, sponsor, participant's study number, name and address of supplier, ‘For Clinical Trial Use Only’ and the dose directions.
Blinding
As this was an open trial, there was no need for procedures to enable study sites to reveal random allocations. Nevertheless we shall keep the CI, statistician and other staff responsible for analysis and reporting blind to treatment allocated and received until they complete primary analysis to the satisfaction of TSC and DMEC.
Interventions
Participants randomised to infliximab received it as remicade in 5 mg/kg intravenous infusions over 2 h—at baseline and 2 and 6 weeks after the first infusion, in accordance with local prescribing guidelines.
Participants randomised to ciclosporin received it as sandimmun by continuous infusion of 2 mg/kg/day. Sites were advised to change the infusion every 6 h using non-polyvinyl chloride bags and administration sets but, given the pragmatic nature of the trial, could follow local practice. Intravenous treatment continued for up to 7 days if successful. Sites switched participants responding to ciclosporin to twice-daily oral doses delivering 5.5 mg/kg/day, and adjusted doses to achieve trough ciclosporin concentration of 100–200 ng/mL. They measured whole-blood ciclosporin levels according to local practice, ideally 48 h after oral therapy and then every 2 weeks. After 12 weeks treatment was at the discretion of the participant's consultant. We asked centres to consult the online Summary of Product Characteristics for remicade or sandimmun and oral ciclosporin at the time of first prescription.
For both treatments we gave centres discretion to start azathioprine or 6-mercaptopurine at therapeutic doses in week 4. We asked them to eliminate steroids by week 12 in patients who remained well but to reinstate them in patients who became symptomatic. We also advised centres to give septrin as prophylaxis against Pneumocystis jiroveci (carinii) pneumonia in both groups.
Outcome measures
Until April 2014, that is between 1 and 3.5 years from randomisation, we shall continue to measure:
-
Primary outcome measure of quality-adjusted survival, weighted by scores on the disease-specific CCQ which we are validating concurrently. The CCQ expands the validated UK IBDQ29 to cover acute illness and the period after colectomy.
Quality of life:
-
2. Disease-specific quality of life, measured by the CCQ.
-
3. Generic quality of life, measured by SF-1230 and EQ-5D31 questionnaires.
Main clinical outcomes:
-
4. Mortality.
-
5. Colectomies, both emergency and planned.
-
6. Readmissions, including those for non-UC-specific causes.
-
7. Adverse events, subdivided into suspected unexpected serious adverse reactions (SUSARs), serious adverse reactions (SARs), serious AEs (SAEs), adverse reactions (ARs) and AE, including all relevant events in 4–6 above.
Incidence of rarer clinical outcomes:
-
8. Malignancies, subdivided between colorectal, other gastrointestinal and other.
-
9. Serious infections, including bacterial infections, pneumonia and abscesses.
-
10. Renal disorders.
-
11. Other new symptoms, especially attributable to treatment.
Disease activity:
-
12. Criteria proposed by Truelove and Witts24: full blood count, inflammatory markers and albumin at baseline and 3, 6, 12, 18, 24, 30 and 36 months.
Economic outcomes:
-
13. NHS costs, measured by a healthcare resource use questionnaire and hospital activity data.
-
14. Patient-borne costs, including number of days off work and travel costs to healthcare.
Qualitative outcomes:
-
15. Patient views of both drugs and their implications.
-
16. Professional views about the drugs, their administration and ease of handling.
Safety monitoring and reporting
Responsibility for ensuring GCP adherence and reporting AEs in accordance with the clinical trial regulations is the delegated responsibility of the PI and the research team at each site, documented in a clinical trial agreement in place with each site.
All AEs are recorded and assessed by the PI or other designated person, using an AE Screening Form to judge the seriousness, causality and expectedness of the event. If uncertain the PI will summarise the event and refer the decision to the CI. Serious AEs, including SUSARS, are reported to the DMEC and an annual safety report, including serious adverse reactions, is submitted to the Medicines and Healthcare products Regulatory Agency (MHRA) and Research Ethics Committee (REC) for Wales.
To help identify AEs, trial participants are given a membership card showing their study ID. They are asked to show this whenever they are seen by a doctor who is not part of the team treating them for UC.
Data collection
We ask all trial participants to complete questionnaires (including CCQ, SF-12 and EQ-5D) at baseline and 3, 6 and 12 months; and to complete them at 18, 24, 30 and 36 months if they reach these timepoints before April 2014.
In addition, we also ask all trial participants who undergo a colectomy or subsequent corrective surgery to complete the postcolectomy version of CCQ on discharge following surgery, and at 4, 8 and 12 weeks after discharge; we have elaborated the original trial design in these ways to strengthen our estimation of quality-adjusted survival, the primary outcome measure for CONSTRUCT.
We collect baseline data for all patients at recruitment to cohort, including:
Sociodemographic data
-
Age, sex, ethnic group and truncated postcodes, which will be used to generate measures of social deprivation (Index of Multiple Deprivation for England, Welsh Index of Multiple Deprivation for Wales, Carstairs’ Deprivation Scores for Scotland, Northern Ireland Multiple Deprivation Measure scores, and Townsend scores for all four countries).
Administrative data
-
B. Admission details
Clinical data
-
C. Disease history including presenting symptom, duration of disease since first diagnosis, previous medical and surgical treatments received, and details of any previous biological, concomitant or steroid therapies.
-
D. Comorbidities in particular cardiorespiratory, liver and renal disease, diabetes and hypertension.
-
E. UC symptoms and signs including duration of symptoms in current episode, stool frequency, blood pressure, pulse and temperature.
-
F. Treatment including type, dose and duration of steroid therapy.
-
G. Pathology results including full blood count, erythrocyte sedimentation rate, C reactive protein, γ-glutamyltransferase, glomerular filtration rate, albumin, liver function tests, urea, creatinine, electrolytes, total cholesterol and total bilirubin.
-
H. Extent of disease and colonic area affected (Montreal classification of IBD32).
-
I. Histopathology results including stool culture results and histological diagnosis.
-
J. Family history of IBD.
-
K. General: height, weight and smoking status.
Quality of life
-
L. Quality of life (QoL) measures. The three QoL measures are administered as one questionnaire as soon as feasible following consent to the cohort; completion of these questionnaires by trial participants must precede randomisation to infliximab or ciclosporin.
Economic evaluation
Cost-utility analysis will use the EQ-5D to estimate quality-adjusted life years (QALYs) and estimated costs from an NHS perspective. Patient-level data on resource use in hospital over the 3.5 years of the study is monitored using GeneCIS and routinely collected data, supplemented where necessary by scrutiny of hospital medical records. These include drugs, drug-related complications, surgery, inpatient stay (in intensive care unit (ICU), high dependency unit (HDU) or ward) and outpatient clinic attendances. NHS resource use outside hospital, including all contacts with health professionals in primary care and the community, is estimated by the CONSTRUCT resource use questionnaire, which is registered on the Database of Instruments for Resource Use Measurement (DIRUM).33 We shall use current UK national prices where available from published sources34 to value these costs; or, if not, from the finance departments of participating sites. Patient-borne costs are estimated by additional questions at baseline, 3, 6 and 12 months, and potentially at 18, 24, 30 and 36 months, but will be reported separately from the cost-utilty analysis.
Qualitative study
We interview participants by telephone to understand their experiences and perceptions of treatment with infliximab or ciclosporin and other therapies for acute severe UC. We invited all participants to show their willingness to be interviewed on the trial consent form. We used purposive quota sampling to identify 12 consenters from each arm of the trial. This yielded a wide representation of therapy, age and sex of patients from a range of centres. We interview them twice—at about 3 and 12 months after they receive treatment.
First interviews follow a structured format to ensure consistency of data collection. The interviews investigate patients’ priorities for their health and well-being, ease of taking the drugs, side effects and response to treatment. Follow-up interviews use a similar schedule but include additional questions to explore what has happened to participants following treatment, including changes over time in their opinions of the treatment, their interactions with healthcare professionals, and their health.
We also interview healthcare professionals, by telephone or face-to-face, to understand their views about the two drugs, their administration and ease of handling. We have recruited 15 doctors and 9 nurses, stratified by number of patients recruited by their sites. These interviews address the ease of handling a range of drugs for UC, aspects of drug provision that might influence professional preference for one drug over another, and impressions of other professional contributions to treatment and care. This is particularly relevant if interviewing consultant and nurse at the same site. This will enable us to explore different professions’ ways of working, in particular in treating and administering drugs for UC, and thus professional interaction and psychological development.
Data management and record keeping
All data acquisition, storage and transmission comply with the Data Protection Act. The study uses the portfolio of standard operating procedures developed and maintained by the Swansea Clinical Trials Unit, WWORTH, and trains staff accordingly, especially in ensuring confidentiality.
We use our own securely hosted, generic clinical information system (GeneCIS) to support data handling and record keeping. Data are held in a remote, professionally managed repository and accessed over a virtual private network via the secure NHS N3 network. The system also supports data validation and facilitates quality assurance, and is backed up comprehensively every working day. We ask all patients to consent to data capture using this system, and to linkage of their electronically held routine data. Other data sources include participants’ medical records, pathology systems and routine data from HES, mortality data from ONS, and primary care systems. Participating sites have access only to identifiable data for those participants under their care, and cannot view any other records.
Aggregated data for analysis by the staff engaged in quantitative reporting are extracted in pseudonymised form. They do not have access to identifiable data and remain blind to treatment allocated and received until primary analysis is complete. In contrast qualitative researchers have access to identifiable data—non-clinical but not clinical. They audiotape and transcribe their qualitative data, and store them securely, identifying them only by the participant's trial number. The Trial Data Manager (TDM) is responsible for maintaining data quality, reporting on that to both TMG and DMEC, and supplying the DMEC with unblinded data in response to formal requests. Hence the TDM has access to all data, and will not contribute to analysis.
Analysis
Missing data
General principles
We shall adopt a consistent approach to missing data relating to effectiveness and cost-effectiveness except where individual outcome measures require variation in that approach. We shall exclude participants without any follow-up data. For each variable, we shall summarise the frequency of missing data, which affects effective sample size and hence statistical power. If there is reason to suspect that data are not missing completely at random (MCAR), the trial statistician and chief investigator will discuss the findings. If not, we shall use appropriate imputation methods to ameliorate the problem of missing data.35
Internal imputation within a questionnaire at specific data collection point
None of the questionnaires has an official algorithm for imputing individual missing answers; fortunately most measures with such algorithms use poor imputation methods. Because all baseline QoL scores and most follow-up scores originate from clinics with nurse assistance if required, there should be few patients with many responses missing. Some of these will have personal reasons for partial non-completion, so it is unlikely that we accurately can impute their answers—conditionally missing at random (MAR).
Nevertheless, to reduce missing data and make good use of available information, we shall impute missing responses within the three QoL measures. If the incidence of missing data is low, we shall make single use of the expectation maximisation (EM) version of the SPSS Missing Value Analysis module. We shall then impute the corresponding scale scores from the relevant instructions for that scale. If the incidence of missing data is high, we shall consider using multiple imputation methods.
External imputation of quality of life scores
If participants are dead at an interview point, we shall set their EQ-5D scores to 0; to avoid introducing outliers, we shall set their other QoL scores to the minimum value observed for that measure in their treatment group.
Otherwise, we shall impute missing summary scale and subscale scores by appropriate regression models from all available values of that score at other data points and the allocated treatment group. We shall consider including extra predictors such as age, gender and whether currently hospitalised, but as they will usually be covariates in the main analysis, they may play only a limited role in predicting missing values.
Statistical analysis
This will address the first three objectives. Reflecting the pragmatic nature of the trial design, the primary analysis will be ‘by treatment allocated’, also known as ‘by intention to treat’. The primary outcome measure will be Quality-Adjusted Survival weighted by scores on the disease-specific CCQ. Important predictors that may affect QoL during follow-up, hence considered as factors or covariates in general linear models (GLM), include:
-
Data collected at baseline during assessment for eligibility for RCT including the sociodemographic variables age, sex, ethnic group, social deprivation (derived from truncated post-codes) and other baseline information collected for the cohort investigation.
-
Data collected after randomisation such as concomitant medical therapy, continuing steroid treatment, and azathioprine or other immunosuppressive therapy.
Study centre and the baseline value of the relevant outcome measure will always be included in models. Otherwise, if any potential factor or covariate has an F value of less than 1 (and so increases SEs of estimates) it will be removed from the model and the data reanalysed. Rare categories in some factors (such as ethnic group) may need to be combined; if so, the categories to be combined will be assessed before examining outcomes, taking account only of observed numbers in each category and the coherence of the new groupings. Age will be treated as a covariate or factor (dichotomised at the observed median) depending on goodness of fit. The treatment by centre interaction will be tested by an F test.
Multiple observations of QoL measures (EQ-5D, SF-12 and CCQ) at different time points will be analysed using the ‘area under the curve’ (AUC). This provides a link with the economic analysis via QALYs, and gives a practical answer relevant to the objectives of the trial. QoL observations will also be analysed as ‘repeated measures’ to test whether findings are sensitive to the choice of AUC as main criterion.
The AUC and GLM analyses will be supplemented by descriptive univariate comparisons of the mean QoL in the two groups at each time point. Events such as mortality and colectomy will be compared descriptively by cross-tabulating the numbers and proportions in each treatment group, with a CI for the relative risk of the event occurring differentially in the two groups. Where data are available on time to these events, an appropriate survival analysis, based on Cox's proportional hazard approach36 will investigate differences by adjusting for the same potential factors and covariates.
Residual diagnostics will be used where analyses assume normality; if the distributions of residuals are markedly non-normal, data transformation or bootstrapping will be considered. Residual analysis will be used to identify outliers; these outliers will be excluded and the data reanalysed. Outcome descriptions, summaries and comparisons will be expressed in accordance with appropriate CONSORT guidelines (http://www.consort-statement.org), including estimates with 95% CIs to summarise two-tailed tests at the 5% significance level.
Health economics analysis
This addresses the fourth and fifth objectives, and comprises cost-utility analyses from an NHS perspective—within trial and in the long term.
Within trial cost-utility analysis
The main effectiveness measure in this analysis will estimate QALYs from EQ-5D scores at baseline and 3, 6, 12, 18, 24, 30 and 36 months for patients who reach these time points in the trial. Costs will be valued using appropriate local and UK national prices where available, and from published sources.34 Specifically the cost of hospitalisation (excluding those for surgical interventions) will be calculated as the number of inpatient days in each setting (ICU, HDU or ward) and relevant daily unit costs. Costs of surgical procedures will be based on the type of surgery multiplied by the most appropriate health-related group reference cost. Drugs will be valued using prices in the British National Formulary.
Mean differential costs between the two treatment groups will be estimated. Since cost data are often skewed, non-parametric bootstrapping will generate 95% CIs around point estimates.
If one treatment is dominant (lower cost, greater effect) then that treatment is unambiguously more cost-effective. If not, results will be reported as an incremental cost-effectiveness ratio (ICER) showing the extra cost of producing one extra QALY. To decide whether the treatment which is clinically more effective offers good value for money, the ICER will be assessed against a range of monetary thresholds representing the maximum the NHS is prepared to pay for an extra QALY (currently £20 000–£30 000). Treatment is deemed cost-effective if the ICER falls below the threshold.37
Inevitably there is uncertainty around estimated costs and effects of medical interventions. Bootstrapping38 and Monte Carlo simulation techniques will be used for trial analyses and modelling studies respectively to generate the sampling distribution of the mean costs and effects, quantifying this uncertainty. Cost-effectiveness acceptability curves (CEACs) will estimate CIs for the ICERs.
Long-term cost-utility analysis
An existing decision-analytical model simulates the progression of hypothetical cohorts of patients with UC receiving the two treatment strategies and tracks lifetime-associated costs and QALYs.39 We shall populate this model with data from our trial and thus enhance the decision-tree model used for the first 12 months. We shall adapt the ensuing Markov model to extrapolate beyond the main period of follow-up and estimate long-term costs and QALYs. Since long-term efficacy of infliximab in UC is unknown, the model considers a 10-year time horizon to explore uncertainty in long-term outcomes. We shall estimate the probability of surgery from trial data for the period until 3 years and thus adjust the life expectancy predicted by the Markov model. Costs and QALYs will be discounted at 3.5% per annum.
The surgery rates used in the model will be based on data from the trial over 36 months. The relationship between transition probabilities and time will be established from trial data using survival analysis. A parametric Weibull model will be employed to model these data and convert event rates into probabilities. A probabilistic sensitivity analysis40 with 10 000 simulations using Bayesian Markov Chain Monte Carlo (MCMC) methods will address the common problem of parameter uncertainty in economic models.
Qualitative analysis
This addresses the last two objectives. We shall investigate patients’ priorities for their health and well-being, their experiences since treatment and their changing opinions of treatment. In parallel we shall explore healthcare professional views of the administration of infliximab and ciclosporin and the choice between them.
Telephone interviews with participants, both patients and professionals, will be recorded and transcribed. Transcripts will undergo rigorous analysis by standard thematic analysis based on the interview schedule and by schematic analysis of whole-page summations of transcripts.41 Thematic analysis is in keeping with a structured approach to data collection, whereby coding is used as the “method for conceptualising research data and classifying it into meaningful and relevant categories for participants in the study.”42
Three researchers will individually code transcripts from first-round interviews with patients as they are completed. When they have coded four interviews each, the researchers will work together to agree a coding structure and develop an analysis framework. The same analysis framework will be followed with all transcripts and data will be prepared for coding, including reading of all transcripts and familiarisation with data. The same process will be followed for the second-round interviews.
Three transcripts (one from a participant given infliximab, one given ciclosporin and one given ciclosporin followed by surgery) will also be taken forward to group analysis sessions for two reasons: to enhance the analysis framework by verifying if aspects are cogent and agreed by a wider group of analysts, and to give those analysing other study datasets an insight into the lives of people suffering from UC. Members of the study team will read the three transcripts and write three one-page overviews of the main features to tell the story of each participant's health and illness. There will then be a group discussion session to see if there is consensus and if the analysis framework is working. This iterative procedure ensures the validity of the analysis framework and its use.
Once agreement is reached and code saturation is achieved across transcripts, the remaining transcripts will be coded by two of the three original qualitative researchers using the adopted analysis framework. The analysis of the professional interviews will broadly follow the same structure.
Further analyses
No interim or subgroup analyses of trial outcomes are planned. We do plan to triangulate findings from different components of the study using the MATRICS approach.43
We hope to follow participants annually for up to 10 years by questionnaire and record linkage of routine data; the data thus obtained will identify future patterns of costs and effects following the treatment received by participants in the trial.
Dissemination
Research governance
The study has EudraCT Number (2008-001968-36) and clinical trial authorisation from the MHRA. It conforms with the Research Governance Frameworks for England,44 Scotland45 and Wales46; the principles of GCP outlined by the International Conference on Harmonisation (http://www.ich.org/); the EU directive 2001/20/EC47; and the Medicines for Human Use (Clinical Trials) Regulations 2004.48
Data sharing
Whilst funded by the NIHR HTA programme, we shall adhere to the MRC-Wellcome Trust data sharing policy and will make data arising from the project available to the scientific community with as few restrictions as feasible. We will retain exclusive use of data throughout the project. Following completion of the project, we shall deposit extensive anonymised data from this project at the University of Essex data archive. We shall encourage data sharing, especially through the Essex data archive.
Publication
In accordance with good practice we have registered CONSTRUCT in a public registry at http://www.controlled-trials.com/isrctn with ISRCTN 22663589. We shall present study findings at national and international conferences and publish as widely as we can in peer-reviewed journals using the CONSORT guidelines.49 We hope to recognise everyone who has worked on the trial.
Discussion
We undertook a feasibility study to refine patient pathways, and the CCQ and economic health resource-use questionnaires. We then conducted a prepilot study to test the recruitment process up to randomisation and ensure that the main components of this trial worked together. Thereafter we used the resulting cohort of some 40 patients to test aspects of study design beyond initial recruitment. Following the prepilot, we piloted the use of the GeneCIS online data collection system, patient recruitment, randomisation and primary data collection. Each centre was asked to recruit and successfully randomise one participant. This was followed by a meeting of investigators to learn from the pilot. As the pilot uncovered no major problems, we have included cohort and trial participants recruited during the pilot period within the main study.
Acknowledgments
The authors thank their collaborators in National Health Service (NHS) Health Boards and Trusts for their continued support for this study.
References
Footnotes
-
Contributors JGW conceived the trial. JGW, IR, WYC, DC, JM, FR, SER, DR, ACS, LT and KT designed the original protocol. MFA, LA, CC, MG, MH, HH and AW contributed to subsequent protocol development. IR leads the statistical aspects of the trial with AW. HH leads patient-reported outcome measurement and analysis with LA. DC leads the economic analysis with MFA. FR leads the qualitative aspects of the trial with ACS and CC. All the authors have contributed to the general conduct of the study as members of the Trial Management Group, commented on drafts of the manuscript and agreed to the final version.
-
Funding This project is funded by the National Institute for Health Research (NIHR) HTA programme (£2,218,111, project number 06/78/03) which monitors study progress and quality through biannual reports. The NIHR HTA programme will review the final report before publication in full in Health Technology Assessment. The NIHR also encourages grant-holders to seek external peer-reviewed publication. The views and opinions expressed are those of the authors and do not necessarily reflect those of the HTA programme, the NIHR, the National Health Service (NHS) or the Department of Health.
-
Role of Sponsor As sponsor, Swansea University monitors legal and financial probity; it delegates monitoring of progress and quality to the local Clinical Trials Unit—the West Wales Organisation for Rigorous Trials in Health (WWORTH).
-
Competing interests None.
-
Ethics approval The Research Ethics Committee for Wales gave ethical approval (Ref 08/MRE09/42) and each participating NHS Health Board or Trust gave NHS Reseach & Development (R&D) approval.
-
Provenance and peer review Not commissioned; peer reviewed for ethical and funding approval prior to submission.
-
Data sharing statement Whilst funded by the NIHR HTA programme, we shall adhere to the Medical Research Council (MRC)-Wellcome Trust data sharing policy and will, make data arising from the project available to the scientific community with as few restrictions as feasible. We will retain exclusive use of data throughout the project. Following completion of the project, we shall deposit extensive anonymised data from this project at the University of Essex data archive. We shall encourage data sharing, expecially though the Essex data archive.