Article Text

Abstract
Introduction The Valsalva manoeuvre (VM) is a recommended first-line physical treatment for patients with re-entrant supraventricular tachycardia (SVT), but is often ineffective in standard practice. A failed VM is typically followed by treatment with intravenous adenosine, which patients often find unpleasant. VM effectiveness might be improved by a modification to posture which exaggerates the manoeuvre's vagal response and reduces the need for further emergency treatment.
Methods and analysis This is a multicentre randomised controlled clinical trial in 10 UK emergency departments (EDs). It compares a standard VM with a modified VM incorporating leg elevation and a supine posture after a standardised strain in stable adult patients presenting to the ED with SVT. The primary outcome measure is return to sinus rhythm on a 12-lead ECG. Secondary outcome measures include the need for treatment with adenosine or other antiarrhythmic treatments and the time patients spend in the ED. We plan to recruit approximately 372 patients, with 80% power to demonstrate an absolute improvement in cardioversion rate of 12%. An improvement of this magnitude through the use of a modified VM would be of significant benefit to patients and healthcare providers, and justify a change to standard practice.
Ethics and dissemination The study has been approved by the South West—Exeter Research Ethics Committee (REC reference 12/SW/0281). The trial will be published in an international peer reviewed journal. Study findings will be sent to the European and International resuscitation councils to inform future revisions of arrhythmia management guidelines.
Results The trial will also be disseminated at international conferences and to patients through the Arrhythmia Alliance, a patient support charity.
Registration The study is registered with Current Controlled Trials (ISRCTN67937027) and has been adopted by the National Institute for Health Research (NIHR) Clinical Research Network.
- Accident & Emergency Medicine
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Strengths and limitations of this study
-
This is the first randomised controlled trial investigating the effects of a postural modification to the Valsalva manoeuvre (VM) to treat patients presenting with supraventricular tachycardia in an emergency department setting.
-
This pragmatic study is designed to detect a meaningful improvement in the efficacy of the VM using a novel and easily achieved modification to the manoeuvre for the benefit of patients.
-
Although blinding of participants and clinicians is not practically possible, participants will not be told which is the ‘new’ technique and blinded analysis of results and independent review of the primary outcome will be undertaken.
-
The study is designed to detect a 12% absolute improvement in cardioversion rate with the modified VM. This effect size was chosen based on the best available evidence and on a magnitude that we felt would be needed to convince clinicians to change practice, but as a result, the study risks being underpowered to detect a smaller difference in efficacy.
Introduction
Supraventricular tachycardia (SVT) is a common heart rhythm disorder with an estimated incidence of 35 episodes per 100 000 persons/year.1 It usually occurs due to the presence of an accessory electrical pathway within the heart, causing re-entrant tachycardia. Episodes can be self-limiting but often recur and attacks are very unpleasant and disruptive to patients’ lives.2 ,3 Patients often come to the emergency department (ED) during attacks: annually about 50 000 attend an ED in the USA.4 In the UK, about 125 000 people are affected and 50–100 patients will attend an average UK ED with SVT each year (unpublished UK regional audit data).
International first-line emergency treatment guidelines for stable SVT advocate attempted cardioversion with a vagal manoeuvre.5 ,6 The Valsalva manoeuvre (VM) is very safe, and the most effective vagal manoeuvre in adults.7 However, it is performed inconsistently8 and has a relatively low (5–20%) cardioversion success rate in clinical practice.9 ,10 Patients who remain in SVT after a VM are usually treated with intravenous adenosine. While effective, this drug causes unpleasant side effects. Some patients report that they feel as though they are about to die; many find this treatment frightening or unpleasant, and significant adverse events have been reported.2 ,11 Other emergency treatments for SVT such as direct current cardioversion are invasive and also not without risk.
Performing the most effective VM will reduce the number of patients presenting in SVT who need to undergo further emergency treatment, and may also increase the number of patients who are able to manage their own condition in the community, without the need to attend hospital.
The efficacy of the VM might be influenced by the duration and intensity of the strain or the position of the patient. Current evidence would suggest that the optimal strain duration is 15 s with a minimum pressure of 40 mm Hg,9 although this strain is not routinely standardised.12 The effect of patient position during emergency VM has not been well studied, but posture modifications to increase venous return and vagal stimulation in the relaxation phase may improve VM efficacy.13–16 There have been no randomised clinical trials assessing the effect of patient position during the VM in ED patients presenting with SVT. Although greater vagal tone is achieved in normal participants with a supine VM,17 it is not known whether this results in a higher cardioversion rate in patients with SVT,13 and the standard VM is typically performed in the semirecumbent position in the ED.8 ,14 ,18
In a non-randomised before and after study, the introduction of a head down (Trendelenburg) position VM into routine practice was associated with an improvement in cardioversion from 5% to 31%.14 This was a very small (19 patient), uncontrolled observational study and its results might not be generalisable to the larger ED population. However, it has been suggested that a further surge in venous return (and thus vagal stimulation) can also be achieved during the relaxation phase, immediately after the Valsalva strain, by laying the patient flat from sitting and lifting the patient's legs (the modified VM).13 ,15 Anecdotally, this modification has often been successful when a standard VM has failed.16 Although the modified VM is based on sound physiological principles and is used by some emergency physicians in our region,18 the effect of this modification on cardioversion rates in ED patients has not been previously studied and is not in widespread global use.
Our study has been designed to determine whether such a modification to the standard VM is more effective in converting SVT to sinus rhythm than the standard sitting VM in stable adult patients presenting to the ED with SVT. A clinically significant improvement in the success rate of the VM using a simple, safe modification to patient positioning during the manoeuvre would be an important discovery, with major benefits for patients and for emergency healthcare providers around the world, and is likely to influence international tachyarrhythmia guidelines.
Methods and analysis
General study design and conduct
This study is a randomised, assessor blind, multicentre parallel group trial in stable adult patients presenting to the UK EDs with SVT. Three hundred and seventy-two participants will be randomised in a 1:1 ratio to undergo a standardised Valsalva (15 s, 40 mm Hg) strain as either a ‘standard’ (control) VM, performed in a semirecumbent position (sitting at 45°), or as a ‘modified’ (intervention) VM, performed semirecumbent but followed immediately by being laid flat with the patient's legs passively raised to 45° for a further 15 s after the strain. This is an open study, but participants’ study allocations will be blinded to those performing a central review of ECGs and the statistician analysing the results.
The key objective of the study was to determine whether a modified VM is better at restoring sinus rhythm (as determined by a 12-lead ECG read by the treating clinician) at 1 min post-manoeuvre in adult patients with SVT compared with the standard VM.
We will also assess the impact of a modified VM on the subsequent need for adenosine and other emergency treatments, on the time patients spend in the ED and on the need for admission to hospital. We will also monitor any side effects or complications of using a modified VM compared to the standard VM. All ECGs will be centrally reviewed by an independent cardiologist to confirm the treating clinician's interpretation.
Patient and public involvement was ensured at all stages of trial design and conduct through patient and patient support group representation on the trial management group, trial steering committee (TSC) and authorship. Day-to-day trial management is administered through the UKCRC-registered Peninsula Clinical Trials Unit (CTU) at Plymouth University and sponsored by the Royal Devon and Exeter National Health Service (NHS) Foundation Trust.
Study procedures
Study population and setting
All adult patients (aged 18 and over), presenting to participating EDs (see Participating hospitals) at any time with suspected SVT, will be considered for inclusion in the trial. Unstable patients with systolic blood pressure less than 90 mm Hg or any indication for immediate cardioversion, those unable to consent or perform the trial VMs and those in whom the trial interventions could conceivably cause harm will be excluded. Full inclusion and exclusion criteria for the trial are summarised in box 1.
REVERT study inclusion and exclusion criteria
Inclusion criteria
-
Age 18 years and older
-
Diagnosis of supraventricular tachycardia (SVT; regular, narrow complex tachycardia with QRS duration <0.12 s on ECG)
Exclusion criteria
-
Unable or unwilling to give informed consent
-
Unstable condition (systolic blood pressure (BP) <90 mm Hg or any indication for immediate drug or direct current cardioversion)
-
Atrial fibrillation or atrial flutter on ECG
-
Suspected atrial flutter requiring a trial of adenosine
-
Severe hypertension (systolic BP>220 mm Hg or diastolic BP>120 mm Hg)
-
Any contraindication to or inability to performing a Valsalva manoeuvre
-
Contraindication or inability to lay flat and have legs lifted (or any reason identified by the patient as to why this manoeuvre would cause discomfort or pain)
-
Third trimester pregnancy
-
Previous inclusion in the study
Screening, recruitment and consent
The screening and recruitment of patients, delivery of the intervention and recording of outcomes will all be carried out within participating UK NHS EDs. It is usual UK practice that patients who come to the ED with palpitations or tachycardia have an ECG performed on arrival, which is reviewed by a senior ED clinician. Patients presenting to the ED with SVT confirmed by an ECG review will be screened for eligibility by Good Clinical Practice (GCP) trained staff. Eligible patients will, by definition, be stable and not in imminent danger; therefore, there will be reasonable time in the ED to obtain informed consent from each participant. Additional checks and reminders will be employed to ensure that all eligible patients have equal opportunity to participate. ED attendance logs will be screened for the duration of the trial to enable the identification of any missed patients with reporting of simple demographic data to ensure that there is no evidence of recruitment bias.
The written informed consent process will normally be undertaken by the attending clinician, but may be delegated to another appropriate member of the research team depending on individual circumstances. Study team members nominated by the principal investigator to undertake the consent process will be listed as such on the study delegation log.
Eligible patients will be provided with an initial summary participant information sheet, a verbal explanation of the purpose and nature of the trial and a description of what participation in the trial will entail. Patients will be given the opportunity to ask questions about the study. Following this verbal and written explanation, patients who are interested and eligible will be invited to participate in the study. If a patient agrees to participate, he/she will be asked to complete an Informed Consent Form which will be countersigned by the staff member taking consent. This will include a signed statement that the patient cannot foresee any reason that the standard or modified VM should cause them any difficulty or discomfort. A record of the patient's consent to participate will be documented in the hospital notes where a copy of the completed consent form and participant information sheet will also be filed.
Baseline assessment and ECG
Study participants will undergo standard clinical assessment in the ED, including medical history, physical examination, recording of routine initial observations of pulse, blood pressure, respiratory rate, oxygen saturation and the recording of a (pre-VM) 12-lead ECG. Baseline data required for the purposes of the trial will be recorded on a study specific case report form (CRF) by the research nurse or nominated research team member. This includes time of arrival in the ED, demographic data, medical history (including previous SVT), current medication, prehospital care (including any VM attempts) and baseline vital signs. All concurrent medications will be recorded on the CRF. There are no concurrent medications which would preclude trial participation.
Randomisation:
After consent has been obtained, participants will be randomised to receive the standard VM (control) or the modified VM (intervention) as the first treatment for their SVT. Treatment allocations will be randomly determined using variable-sized blocking and stratified by centre. The randomisation sequence will be prepared by an independent statistician using proprietary software, and then passed to the CTU for preparation of allocation cards. These cards will be placed in sequentially numbered opaque sealed envelopes, independently and in advance, by the CTU and provided to participating sites.
These envelopes will be tested for 100% concealment and closed with a tamper-proof seal, preventing opening and resealing. Envelopes will be securely stored in a locked ED cupboard at each site, access to which is controlled by a senior nurse, who is independent to the study but always present when potentially eligible patients attend the ED. Envelopes will be sequentially released by the CTU in small batches sufficient to support projected recruitment and replace used envelopes. Envelopes will only be accessed after participant consent and with the agreement of the recruiting doctor and resuscitation room nurse.
The treatment will be allocated by selecting the next presealed randomisation envelope in sequence. The number on the envelope will be checked against a randomisation log to ensure that the correct envelope has been selected. The recruiting doctor or nurse will complete the randomisation log each time an envelope is taken for use and will sign and date the envelope across the unbroken seal to confirm that the next available and lowest numbered envelope of the batch has been taken, that it is unopened and bears no evidence of tampering. The participant's initials and date of birth will also be written on the envelope prior to opening. Correct and sequential use of envelopes will be strictly audited by the site research team and CTU trial manager.
Trial interventions
Consenting, eligible patients will be randomised to the standard (control) or modified (intervention) VM, using standardised manoeuvres according to instructions and training provided specifically for the trial.
Participants in both groups will be placed in a semirecumbent position, sitting at an angle of 45°, for at least 1 min prior to undertaking the VM. All patients will have standard monitoring including a continuous 3-lead ECG, non-invasive blood pressure monitoring and oxygen saturation monitoring during any VM.
Standard VM (control)
Participants will be seated at a 45° angle, and will perform the standardised strain for 15 s and remain in this seated position for 60 s. To reduce bias, this allocation will be described to patients as the ‘stay sitting Valsalva’.
Modified VM (intervention)
Participants will be seated at a 45° angle, and will perform the standardised strain for 15 s. Following this, they will be laid flat, and their legs will be raised by ED staff to a 45° angle for 15 s. Participants will then be returned to the 45° semirecumbent position for 45 s. To reduce bias, this allocation will be described to patients as the ‘lying down with leg lift Valsalva’.
Standardised strain
Participants in both groups will be instructed to perform a standardised strain (VM) for which instructions are provided specifically for the trial and printed on the reverse of each allocation card. This consists of a forced expiration against a manometer (Welch Allyn) to a pressure of 40 mm Hg for 15 s, through a disposable 3 mm green (bubble) oxygen tubing (Uhs Ref:UN881) cut at its widest point for the mouth end and at 92 cm to allow connection to the meter. The meter, marked with the target pressure and a timer, will be visible to the participant, the treating clinician and attending nurse. During the strain, clinicians will give standardised verbal encouragement at 5 and 10 s to support participants in achieving target pressure and strain duration. A stopwatch is attached to the manometer for consistent timings. If sinus rhythm is not restored, according to a 3-lead ECG monitoring, patients in both groups will be invited to undertake one further VM of the allocated type.
The clinician will record on the reverse of the allocation card whether the allocated VM was attempted and the peak pressure and the total duration of strain achieved. The duration of the longest strain will be recorded if more than one attempt was needed during the 15 s period. This information will be used to report whether the Valsalva was performed correctly, though since statistical analyses will be on an intention to treat basis, participants performing the manoeuvre incorrectly will not be excluded from the study. The attending clinician or nurse will also ask the participant if the VM caused any discomfort or problems and will record the response together with any technical problems or clinical adverse effects that occurred during the VM.
Postintervention management
Patients will be left in position for 1 min after the VM before undergoing a repeat (post-VM) 12-lead ECG. This will ensure that very transient non-sustained return to sinus rhythm during the manoeuvre, which can occur, is not recorded as a treatment success. Individual trial participation will last from the time of patient consent until the recording of this postintervention (Valsalva) ECG (figure 1). Typically, this process will take less than 1 h.

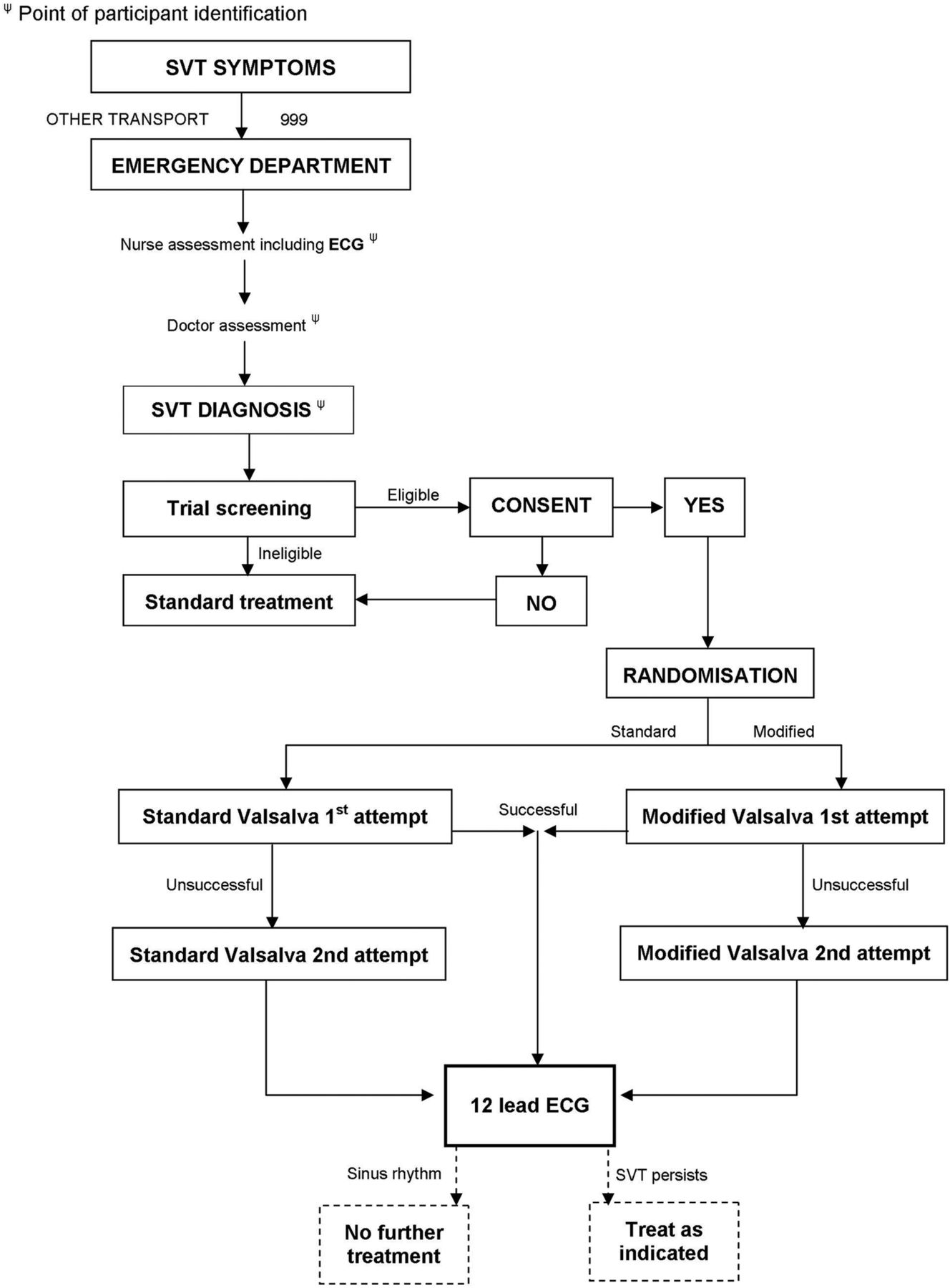{kind=link}
REVERT study flow diagram.
Subsequent management of the patient will be entirely according to standard clinical guidelines with no further trial intervention. Participants will be followed up until ED discharge or hospital admission with details of any further ED treatments for SVT recorded. The time of ED discharge will be verified from the patient tracking system and recorded in the CRF. For the purposes of the study, ED discharge is defined as discharge to an inpatient or observation ward or discharge out of the hospital. Time spent in an ED observation ward, for example, awaiting transport home, will not be included in the total time spent in the ED.
Prior to discharge, all participants will be given a more detailed information sheet about the study, including written instructions on how to perform both types of VM themselves once they have left hospital. An equivalent Valsalva strain can be achieved by blowing into a 10 mL syringe with just enough force to move the plunger and an unused syringe will also be given to all trial patients to take away.19 The information sheet will include a weblink to the Arrhythmia Alliance website (http://www.arrhythmiaalliance.org.uk), where information will be posted at the end of the trial explaining which manoeuvre has been shown to be most effective.
Independent ECG review
Copies of pre-VM and post-VM ECGs will be made and labelled with the participant's initials and trial number to preserve anonymity. All ECGs will be reviewed by an independent assessor (consultant cardiologist) blind to treatment allocation and not a member of the research team. Any disagreement when compared with the clinically recorded assessment, or any other uncertainty in ECG diagnosis, will be arbitrated by a consultant cardiology electrophysiologist blind to group allocation.
Safety monitoring
The VM is a safe, standard recommended intervention and the trial has no separate data safety monitoring committee. However, the trial exclusion criteria were designed to exclude patients in whom either trial VM could conceivably cause harm and staff have been trained to document and report all adverse events occuring at any point during trial participation. All suspected serious adverse events (SAEs) will be reported, within 24 h of discovery, to the CTU who will notify the chief investigator, the TSC and the trial sponsor. All SAEs will be followed up until resolution.
A summary report of all serious and non-SAEs will be scrutinised by members of the TSC on a quarterly basis. The TSC and trial sponsor have the authority to stop the trial if any indication of harm is found. The trial will be fully compliant with GCP guideline standards for adverse event reporting.
Hypothesis and effect size
The null hypothesis for the trial is that there is no difference in cardioversion rates of SVT between the standard and modified VM. Given the availability of an effective, albeit unpleasant, drug treatment for SVT, we felt that a clinically significant difference between the two groups would need to be at least in the magnitude of 10–15 percentage points to persuade clinicians to change their practice. This effect size, while large, is substantially less than that reported in the best currently available evidence (absolute improvement of 26% seen in a non-randomised trial).14
Primary outcome measure
The primary outcome for the study will be the presence of sinus rhythm as determined by the 12-lead ECG performed 1 min after the Valsalva attempt, read by the treating clinician.
Secondary outcome measures
Secondary outcomes will include: the use of adenosine (including doses and number of treatments), the use of other ED treatments for SVT, the need and reason for admission to hospital, as documented in the hospital notes, and the length of time participants spent in the ED, as recorded by the hospital electronic patient tracking systems. The number and nature of any adverse events and the confirmation of the cardiac rhythm on the post-Valsalva ECG, as determined by a consultant cardiologist (arbitrated by an electrophysiologist in the event of disagreement with the treating clinician's interpretation), will also be reported.
Determination of sample size
Local audit data and previous ED studies suggest a standard VM baseline success rate of about 15%. To demonstrate a 12% absolute improvement from this baseline to a 27% cardioversion rate with a modified VM will require an estimated 186 patients per group. This calculation assumes a two-tailed test of statistical significance with an α level of 0.05 and power of 0.8.
To achieve this sample size, we estimated that a 22-month recruitment period across at least nine centres would be needed. This is based on a conservative mean attendance rate of 1.8 adult SVT patients per week seen in local audit and a recruitment rate of around 25% of eligible adult patients attending with SVT, after excluding 10% of patients who had one or more trial exclusion criteria (unpublished audit data). As the trial participation period is so short, study dropout would be expected to be restricted to participants withdrawing consent prior to or during the intervention, or participants with spontaneous cardioversion between recruitment and trial intervention. We expect rates of these to be very low, and so we have not included any extra participants in the sample size calculations to compensate for this, but dropout rates will be carefully monitored and recruitment increased through the addition of further recruiting centres if needed to ensure that the planned sample size is achieved.
Data analysis
A detailed statistical analysis plan, detailing the exact analyses to be conducted and reported, will be developed prior to any comparative analysis. All analyses will be conducted using a database containing codes for group allocation without indicating which specific group each code refers to. A CONSORT20 diagram will be used to report numbers of patients screened, recruited and randomised. It will also detail numbers receiving each procedure and any dropouts.
There is no opportunity for cross-over of trial treatments except in the case of error. If such an error occurs, participants will be analysed according to their trial allocation on an intention-to-treat basis.
Baseline data (eg, demographics and medication) will be reported descriptively by group. All comparative analyses will be conducted in accordance with intention-to-treat principles and reported with suitable point estimates, 95% CIs and p values. Sensitivity analyses will be conducted for any missing data or protocol violations. Rates of successful cardioversion to sinus rhythm will be compared using a logistic regression with allocation group and centre as fixed effects. Similar analyses will be conducted for receipt of further treatments and hospital admission. Time spent in the ED will be compared using time-to-event methods, namely a Cox Proportional Hazards model (including centre).
We considered collecting prehospital data on duration of symptoms of SVT and prior (eg, patient or paramedic) VM attempts. These data, however, are not routinely, accurately or reliably recorded and do not usually influence ED management or preclude use of a VM as first-line ED treatment. Therefore, to maintain the trial's simplicity and pragmatic approach, we did not design the study to collect such prehospital information. Although prior VM attempts or duration of SVT could potentially act as confounders, we felt that randomisation and allocation concealment would sufficiently control this potential allocation and recruitment bias.
We also considered, but decided against, an analysis of the relative frequency of SVT subtypes, for example, atrioventricular nodal re-entry tachycardia and atrioventricular nodal re-entry tachycardia (AVNRT) in participants. This is not a subdiagnosis routinely made by treating emergency physicians; nor is it one which influences SVT treatment in the ED. Furthermore, the data being collected (pre-Valsalva and post-Valsalva ECGs) would not alone allow reliable retrospective subtyping of the ECGs. No interim analyses are planned.
Missing data
Given the nature of the short follow-up and limited amount of data to be collected, it is not anticipated that there will be a significant quantity of missing data. Every attempt will be made to retrieve any missing data. Where there remain missing data, sensitivity analyses may be conducted using a range of assumptions where necessary, and consideration given to suitable imputation.
Data monitoring
Although there is no formal data monitoring committee, rates of overall cardioversion and adverse event data will be monitored by the TSC to ensure safety. Rates of postrandomisation (pre-Valsalva) spontaneous cardioversion will be monitored, as spontaneous cardioversion does occur in SVT. Numbers of patients recruited whose final or expert presenting ECG diagnosis was other than SVT (eg, some cases of atrial flutter can be misinterpreted as a re-entrant SVT) will also be monitored. Although these cases will still be analysed on an intention-to-treat basis under their original treatment allocations, additional patients will be recruited to ensure that there are sufficient numbers of genuine SVT cases undergoing trial intervention to meet the prespecified sample size calculations.
Ethics and dissemination
The trial complies with the Declaration of Helsinki and GCP guidelines. All eligible, willing participants will undergo written informed consent by GCP trained staff before taking part in the study.
The results of the study will be applicable and of interest to emergency physicians, paramedics, acute physicians, general practitioners and patients. We aim to disseminate the findings of the study to these groups through publication of the trial in an appropriate international peer reviewed journal and through presentations at local, national and international academic meetings such as the College of Emergency Medicine scientific meeting and International Conference of Emergency Medicine. Chairs of the UK, European and International resuscitation councils will also be informed of the results in order to inform subsequent guidelines for the acute management of SVT.
All patients recruited into the study will be offered an instruction sheet detailing how to perform both VMs themselves, and where to find out the results of the trial through the Arrhythmia Alliance and its website. The results will also be accessible to people with SVT outside the study through this patient support organisation. At the conclusion of the study, this website will also provide guides for patients and professionals on how to perform the most effective VM, both in and out of hospital. Links to this resource will be sent to administrators of established online medical education sites which include the management of cardiovascular emergencies.
Discussion
The Randomised Evaluation of modified Valsalva Effectiveness in Re-entrant Tachycardias (REVERT) trial is the first randomised controlled trial to assess the effect of modification to the VM in the treatment of patients presenting to the ED with SVT. It aims to discover whether a simple postural modification to the standard VM is more effective in returning patients to sinus rhythm, and reducing the number of patients who need to proceed to unpleasant drug treatment. This would have potential benefits for patients around the world who suffer from SVT both in and out of hospital as this treatment can be performed by the patient themselves without specialist drugs or equipment.
The trial has been designed with patients in mind, involved patients throughout the process and addresses a frequently occurring problem. It is a pragmatic study carried out in the environment where these patients are normally treated, and where the results will be applied. The study is based on routine current treatment, with appropriate modifications to ensure robust trial methodology, and complies with guidelines for randomised trials.20 The trial has also been specifically designed to meet its objectives and recruitment targets in the challenging ED environment using a carefully considered effect size and accrual rate, based on data from participating sites and previous research. We believe the trial will therefore be able to deliver these objectives within the allocated time and resources. The proposed modifications to the VM do not interfere with or replace specialist treatment or referral plans, or impact on long-term treatment options such as accessory pathway radiofrequency ablation.
Benefits of demonstrating an improved VM would include the reduced need for treatments which patients find unpleasant, such as intravenous adenosine, and may reduce the time patients spend in the ED. We have also built-in processes to empower patients to try these techniques out of hospital. A better VM for use by patients themselves would give them more control and may reduce the need for hospital attendance. If the trial shows that the modified VM is more effective, the challenge will be the dissemination of the technique to patients as well as staff. Collaboration with patient support groups in the delivery of this trial is an important part of its design, and lays the foundations to meet this challenge.
Conclusions
The REVERT trial will seek to determine whether a simple postural modification to the VM improves cardioversion rates in adult patients presenting to the ED with SVT. It is an important trial for people with SVT and the healthcare staff who are involved in their care. The results of the study could lead to a significantly improved VM and reduce the need for patients to be treated with more invasive treatments during attacks, saving time in the ED and potentially avoiding the need to attend hospital altogether, if successfully employed by patients themselves.
Acknowledgments
The authors would like to acknowledge and thank Corinna Phillips, Peninsula Clinical Trials Unit, for all her hard work and administrative skills in support of this project and Paula Garrett, our patient representative, for her help and advice in the ethics application and her views on the protocol and research plans. They would also like to thank Mark Dayer and Andrew Sharp for their expert and independent ECG review for the study.
References
Footnotes
-
Contributors AA conceived the idea for the study. AA, AR, CM were responsible for the initial study design, which was refined with the help of PE, AB, JB and JG and support of our regional National Institute for Health Research (NIHR) Research Design Service. PE provided the statistical plan and JG the background information and audit data for the trial. TL represented the public and patient views in the study development. The Heart Charity, which TL leads, provided patient testimonies that helped inspire the trial design. All authors contributed to the final study design and protocol development, critically revised successive drafts of the manuscript and approved the final version. The trial management group is responsible for the conduct of the study, overseen by a Trial Steering Committee.
-
Funding This research project is funded by the National Institute for Health Research (NIHR) through its Research for Patient Benefit (RfPB) Programme (Grant Reference Number PB-PG-0211-24145).
-
Competing interests None.
-
Ethics approval The study has been approved by the South West—Exeter Research Ethics Committee (REC reference 12/SW/0281) and each of the participating NHS Trust's Research and Development departments.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) licence, which permits others to distribute, remix, adapt, build on this work non-commercially, and licence their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. http://creativecommons.org/licenses/by-nc/3.0/.