Article Text

Abstract
Introduction Major depression is a serious mental disorder that causes substantial distress and impairment in individuals and places an enormous burden on society. Although antidepressant treatment is the most common therapy provided in routine practice, there is little evidence to guide second-line therapy for patients who have failed to respond to antidepressants. The aim of this paper is to describe the study protocol for a randomised controlled trial that measures the clinical effectiveness of cognitive behavioural therapy (CBT) as an augmentation strategy to treat patients with non-psychotic major depression identified as suboptimal responders to usual depression care.
Methods and analysis The current study is a 16-week assessor-blinded randomised, parallel-groups superiority trial with 12-month follow-up at an outpatient clinic as part of usual depression care. Patients aged 20–65 years with Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM-IV) Major Depressive Disorder who have experienced at least one failed trial of antidepressants as part of usual depression care, will be randomly assigned to receive CBT plus treatment as usual, or treatment as usual alone. The primary outcome is the change in clinician-rated 17-item GRID-Hamilton Depression Rating Scale (GRID-HAMD) score at 16 weeks, and secondary outcomes include severity and change in scores of subjective depression symptoms, proportion of responders and remitters, safety and quality of life. The primary population will be the intention-to-treat patients.
Ethics and dissemination All protocols and the informed consent form comply with the Ethics Guideline for Clinical Research (Japanese Ministry of Health, Labour and Welfare). Ethics review committees at the Keio University School of Medicine and the Sakuragaoka Memorial Hospital approved the study protocol. The results of the study will be disseminated at several research conferences and as published articles in peer-reviewed journals. The study will be implemented and reported in line with the CONSORT statement.
Trial registration number UMIN Clinical Trials Registry: UMIN000001218.
- major depressive disorder
- cognitive behavior therapy
- randomized controlled trial
- clinical protocols
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Strengths and limitations of this study
-
This protocol will provide new evidence concerning the administration of cognitive behavioural therapy (CBT) for major depression as an augmentation strategy for patients who have failed to respond to pharmacotherapy in psychiatric care settings.
-
Central randomisation and blinded assessment will be used.
-
The study cannot examine the efficacy of CBT itself because there no attention-placebo control group.
-
The small number of study sites may affect generalisability.
Introduction
As in other high-income countries, major depression is a common mental disorder in Japan.1 Left untreated, major depression can cause substantial distress and impairment in individuals and negatively affect their quality of life, morbidity and mortality; it also places an enormous burden on society.2–4 Latest estimates from the Global Burden of Disease (GBD) 2010 study indicate that major depression accounts for 2.5% of the global disease burden,5 and it is predicted to be the leading cause of disability in high-income countries by 2030.6 Treatment guidelines for this debilitating mental disorder recommend antidepressants for first-line treatment of moderate to severe acute major depression,7 ,8 and this remains the most common treatment in routine practice. However, available evidence indicates that only a third of patients fully respond to initial treatment with antidepressants.9–11 Thus, many patients with major depression are left with considerable symptomatology after initial treatment and are described as having treatment resistant (refractory) depression (TRD).
Although many treatment studies investigating the best treatment strategies for TRD have been reported,12 ,13 no standard treatment has yet been established.14 When patients fail to respond to an adequate course of antidepressants, current guidelines recommend increasing the dose of the current antidepressant, switching to a different antidepressant, or augmenting treatment with other drugs.7 However, one major problem with TRD is the lack of agreed operational definitions.15 ,16 Given the heterogeneity of TRD associated with complex aetiological pathways, a non-pharmacological approach such as depression-specific psychotherapy may have a role in treatment.17
It is well established that cognitive behavioural therapy (CBT), the best known structured form of psychotherapy and which is based on Beck's cognitive theory,18 is effective for the treatment of depression.7 ,8 Numerous randomised controlled studies have shown that CBT is superior to wait-list, non-specific controls or treatment as usual.19 Further evidence shows that combining psychotherapy with pharmacotherapy is more effective than pharmacotherapy alone.20 As a result, CBT has been gaining considerable attention in Japan as an effective treatment for depression among clinicians, academics and the general public. The Japanese Ministry of Health, Labour and Welfare has been encouraging training for, and practical implementation of, CBT, as exemplified by the coverage of CBT for mood disorders by the Japanese national health insurance scheme since 2010.21
Despite these developments, few empirical studies have evaluated the effectiveness of CBT as a second-line therapy for patients who have failed to respond to antidepressants.22–24 The Sequenced Treatment Alternatives to Relieve Depression (STAR*D) Study examined CBT and pharmacotherapy as a sequential approach to manage patients who had failed an initial 12–14 weeks of citalopram treatment by using either augmentation or switch strategies.25 No differences in outcome after treatment were observed between patients receiving augmentation with CBT and augmentation with other pharmacotherapy, or between patients switching drugs. However, the STAR*D trial implemented a unique equipoise-stratified randomisation design which allowed participants to refuse the non-preferred treatment arm; only a quarter of the STAR*D participants were randomised to CBT for their second-line treatment and this selection bias makes it difficult to interpret the outcomes. Next, Kennedy et al26 compared cognitive therapy and lithium augmentation as a sequential treatment option for 44 outpatients with major depression who had a partial response during 8–14 weeks of antidepressant treatment in an 8-week randomised controlled trial. They found that cognitive therapy showed no significant benefit over lithium augmentation. However, the sample size was small, which may have limited power to detect differences over time, and the duration of the trial was relatively short. Furthermore, the trial focused on partial responders (defined as having a Hamilton Depression Rating Scale (HAMD) score of 8–15) and excluded non-responders to the initial antidepressant treatment. Finally, the recent CoBalT trial27 examined the effectiveness of CBT as second-line therapy for patients whose depression did not respond to usual depression care delivered by general practitioners in the UK. In this pragmatic clinical trial with a sample size of 469, adding CBT to usual care increased the treatment response threefold at 6 months compared to those with usual care alone. However, the primary outcome of this trial was a self-reported measure (ie, the Beck Depression Inventory-Second Edition) that might have been affected by the treatment process. Further, it is unclear if this result could be applied to different clinical settings such as psychiatric care or other socio-cultural contexts.
There is little evidence to guide second-line therapy for patients who have failed to respond to antidepressants in psychiatric care settings. We therefore planned to carry out a randomised controlled trial to examine the effectiveness of CBT as an augmentation strategy for antidepressant non-responders compared with pharmacotherapy as part of usual patient care. The aim of this paper is to describe the study protocol of the current study.
Objectives
The primary objective of this study is to compare the effectiveness of CBT as an augmentation strategy to treatment as usual (which includes treatment with antidepressants) versus only treatment as usual in a 16-week randomised controlled trial with a 12-month follow-up for patients with non-psychotic major depression who were identified as suboptimal responders to usual depression care.
The secondary objective of the study is to evaluate the safety (incidence of treatment discontinuation and adverse events) of CBT as an augmentation strategy to treatment as usual for patients with non-psychotic major depression who have not adequately responded to usual depression care.
Methods and analysis
Study design and setting
The current study is a 16-week assessor-blinded, randomised, controlled superiority trial of two parallel groups with 12-month follow-up at an outpatient clinic as part of usual depression care (figure 1). Random allocation to treatment will be done at the individual level.
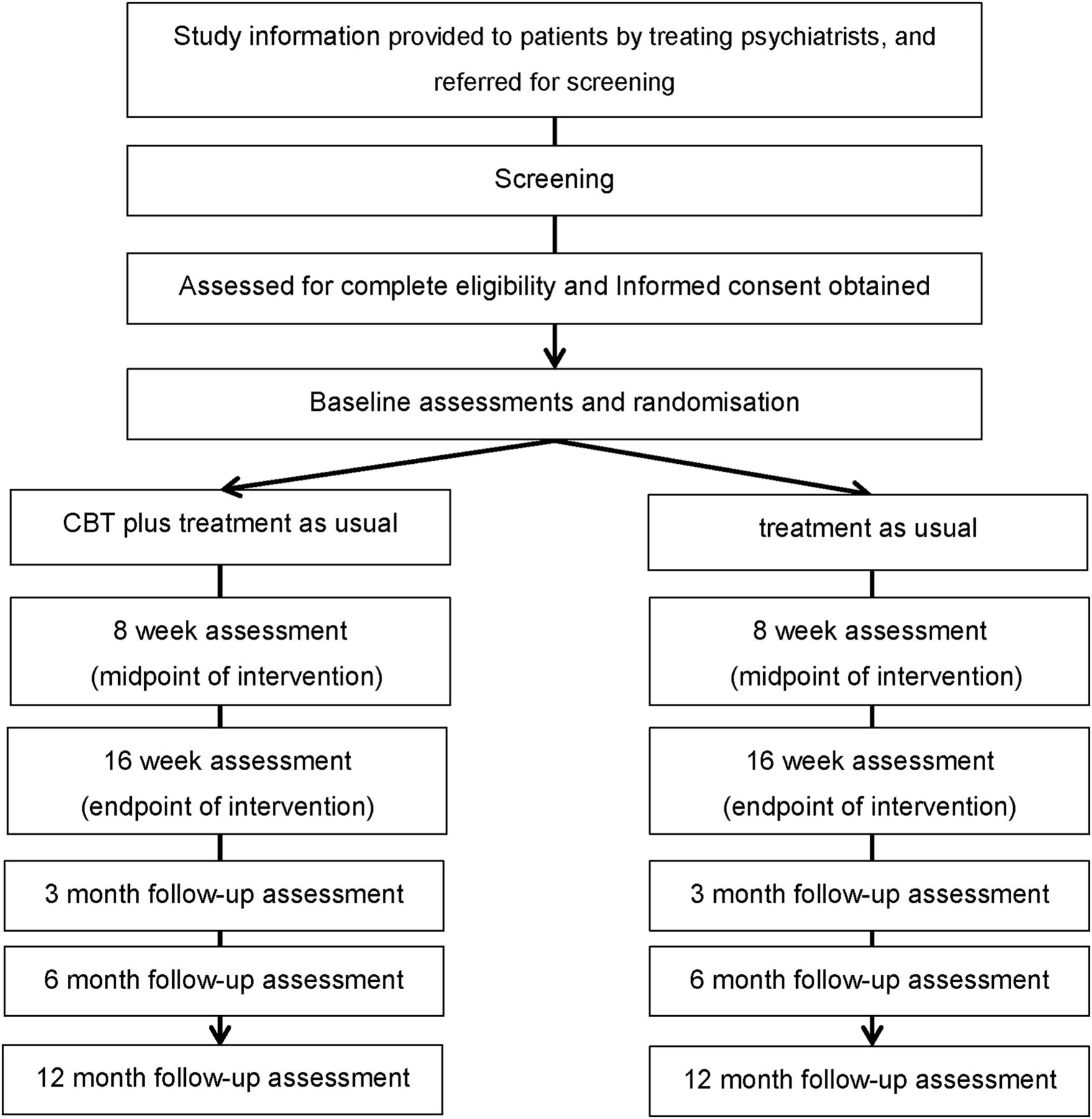
{kind=link}
Implementation of the Effectiveness of Cognitive Behavioural Therapy Augmentation in Major Depression Treatment study (ECAM study). CBT, cognitive behavioural therapy.
Patients will be recruited from two sites in Tokyo. One will be a university teaching hospital, and the other a psychiatric hospital. The university teaching hospital department of psychiatry located in central Tokyo has 31 beds (there are 1044 beds in the entire hospital) and offers advanced psychiatric care services for patients with complex problems, who are largely middle-class Japanese. In contrast, the psychiatric hospital located in suburban Tokyo has 467 beds and offers a wide range of psychiatric care services, mainly providing secondary to tertiary psychiatric care to a diverse Japanese population that includes socioeconomically disadvantaged groups. A feature of Japan's healthcare service is its universal health insurance system whereby all patients receive free access to specialised medical services at all institutions including university teaching hospitals.
Participants
Inclusion criteria
Patients are eligible to be included in the study if they meet the following criteria: (1) they are outpatients with a diagnosis of Major Depressive Disorder, as defined by the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM-IV)28 criteria for single or recurrent depression without psychotic features assessed with the Structured Clinical Interview for DSM-IV (SCID)29 administered by a trained psychiatrist; (2) they are between 20 and 65 years of age; (3) they are suboptimal responders to usual depression care, defined as those who experience at least a moderate level of depression symptoms based on at least 16 items of the 17-item GRID-Hamilton Depression Rating Scale (GRID-HAMD17)30 ,31 and evidence of at least a minimal level of treatment resistance (scoring at least 3 on the Maudsley Staging Method for treatment-resistant depression32) despite receiving antidepressant treatment at an adequate therapeutic dose (based on package insert; see: http://www.info.pmda.go.jp/info/iyaku_index.html) for at least 8 weeks as part of usual depression care; and (4) they are competent and able to give informed consent.
Exclusion criteria
Patients will be excluded from the study if: (1) they have a past or current manic or psychotic episode; (2) they have comorbid alcohol or substance use disorder in the 6 months prior to study entry; (3) they have a primary diagnosis as assessed by the Mini-International Neuropsychiatric Interview (M.I.N.I.)33 ,34 of any DSM-IV Axis I disorder other than Major Depressive Disorder; (4) they have antisocial personality disorder; (5) they have serious and imminent suicidal ideation; (6) they have a serious or unstable medical illness; (7) they have organic brain lesions or major cognitive deficits in the year prior to study entry; (8) they have previously completed a CBT programme; or (9) they are unlikely to attend more than 8 visits during the 16-week trial phase (eg, due to relocation).
Procedures
Recruitment
The treating psychiatrist will, during their usual consultation, provide brief information about the study using a brochure and invite the patient to take part in the study. If the patient shows interest in the study and provides contact details to the research team, a face-to-face appointment with a study psychiatrist will be arranged. The details of the study and its potential benefits as well as risks will be explained thoroughly to the patient by the study psychiatrist and discussed. If the patient agrees to study participation, written informed consent will be obtained, after which the patient will be assessed for eligibility by the study psychiatrist. The SCID29 will be used to confirm a diagnosis of Major Depressive Disorder, while other Axis I disorders will be evaluated with the M.I.N.I.33 ,34 Of note, Axis II disorders will be evaluated with the SCID Axis II Disorders (SCID-II)35 at week 8 (ie, thus allowing plenty of time for depressive symptoms to abate). The diagnostic interviewers will be the study psychiatrists (AN and MS) who have received extensive training in the administration of semi-structured interviews.
Baseline assessment
Acute psychopathology will be assessed at study entry by the study psychiatrists or psychologists. Objective depressive symptoms will be assessed with the 17-item and 21-item GRID-HAMD. Patients’ subjective perception of depression severity will be assessed with the self-reported Beck Depression Inventory-Second Edition (BDI-II)36 ,37 and the 16-item Quick Inventory of Depressive Symptomatology Self-Reported (QIDS-SR16).38 ,39 Health-related quality of life will be measured with the European Quality of Life Questionnaire–5 Dimensions (EQ-5D)40 ,41 and the 36-Item Short-Form Health Survey (SF-36).42 Work performance and productivity will be measured with the WHO Health and Work Performance Questionnaire (HPQ).43 ,44 Life stressors will be measured using the St. Paul-Ramsey Questionnaire (available from the authors), which rates the severity of individual stressors from 1 (none) to 7 (catastrophic) in six categories ranging from marital to occupational, and provides a final global measure of the stressors.
Demographic and other clinical data will be also collected as a part of the baseline assessment, such as marital status, number of children, residential status, level of education, duration of current and lifetime episodes of depression, number of lifetime depression episodes, history of depression treatment including past pharmacotherapy and hospitalised treatment, past suicide attempt, history of medical complications, and family psychiatric history. Level of treatment resistance will be evaluated operationally with the Maudsley Staging Method for TRD.45 A history of childhood abuse and traumatic brain injury will be rated as present or absent. Current cigarette and alcohol use will be determined based on self-report. Assessments will also be conducted at 8 and 16 weeks after randomisation.
Randomisation
All eligible patients who consent to participation will be randomised to treatment as usual or to CBT plus treatment as usual at the end of baseline assessment (1:1 allocation ratio). Randomisation will be conducted using a central computerised registration system designed for this study by the Project Management Office at the Keio Center for Clinical Research. This system automatically randomises patients and generates a message noting their assigned treatment. Allocation will be concealed and stratified by site (n=2) with the minimisation method to balance the age of the participants at study entry (older vs younger) and baseline GRID-HAMD17 score (low vs high). The cut-off age and GRID-HAMD17 score for minimisation will not be disclosed until study completion to ensure concealment.
Intervention phase (16 week)
CBT
Therapists will follow the CBT treatment manual for depression developed by the authors (YO, DF, AN, TK and MS) (available at the Japanese Ministry of Health, Labour and Welfare website: http://www.mhlw.go.jp/bunya/shougaihoken/kokoro/dl/01.pdf). This manual is based on Beck's treatment manual46 with some adaptations in order to address the cultural characteristics of Japanese patients, such as greater emphasis on interpersonal relationships and consideration of the family as an essential part of treatment.47 An overview of the programme is shown in table 1. Problem-solving techniques and specific approaches to address interpersonal issues and cognitive behavioural avoidance are emphasised. Therapists are encouraged to refer to the relevant approaches when necessary. Furthermore, the therapists are encouraged to give feedback to the patients about case conceptualisation and collaboratively set the treatment goal during early in the programme. The patients allocated to CBT will typically receive a course of 16 weekly sessions, with up to four additional sessions if deemed clinically appropriate by the study therapist (maximum of 20 sessions, and minimum of 8 sessions). Sessions will last approximately 50 min. Therapy will take place in an outpatient consultation room at each site. Other depression-specific empirical psychotherapy (ie, interpersonal therapy, IPT) and electroconvulsive therapy are not allowed during CBT treatment.
Framework of the 16 weekly sessions delivered during the cognitive behavioural therapy (CBT) programme for depression
Training and supervision of therapists
Six therapists will deliver CBT at the two sites. The study therapists are trained psychiatrists (n=4), a master's degree clinical psychologist (n=1) and a psychiatric nurse (n=1). Two of the six therapists are female (33.3%). On average, the study therapists will have 4.0 (SD 2.1) years of experience as CBT therapists and will have completed 12.5 (SD 7.3) cases at the time of participation. All therapists have received CBT training under the Keio University Cognitive Behavioural Therapy Training and Research Program and will receive continuous supervision throughout the study.
To ensure treatment fidelity, all therapists will have completed a 2-day workshop and will participate in 2 h group supervision sessions that are held once every two weeks with other therapists during the study. Therapists will present case formulation and treatment plans at the group supervision sessions, which will be led by one of the authors (YO), the founder and the president of the Japanese Association for Cognitive Therapy and a fellow of the Academy of Cognitive Therapy, who will facilitate discussion of therapeutic difficulties and lack of progress, aid skills acquisition, and provide peer support. To assess therapist competence in CBT, a random sample of audiotaped sessions will be rated using the Cognitive Therapy Rating Scale (CTRS).46 ,48 A score of 40 or greater is defined as an adequate level of technical competency in CBT.
Treatment as usual (usual depression care by psychiatrists)
Although appropriate flexibility will be allowed for scheduling sessions, the patients will typically receive a 5–30 min consultation once every two weeks by the treating psychiatrist during the treatment phase with a minimum of eight sessions. A typical session will consist of symptom assessment and standard clinical management such as brief psychoeducation and pharmacotherapy when appropriate. Although there will be no restriction on pharmacotherapy, it should basically comply with practice guidelines for major depression such as those of the American Psychiatric Association.8 Prescribed medicines and doses will be recorded and medication adherence will be assessed at each visit using the self-reported Treatment and Medication Compliance Data Scale (TMCDS) (available from the authors upon request). Study participants are allowed to receive medication for concurrent general medical conditions. No depression-specific empirical psychotherapies (CBT or IPT) or electroconvulsive therapy are permitted during the intervention phase and will result in withdrawal from the study. Treatment will be delivered by seven treating psychiatrists who have practiced for a mean of 7.3 (SD 4.4) years and are working at the two sites.
Follow-up phase
There will be no restrictions on treatment options for the patients who receive depression care from the treating psychiatrists during the follow-up phase. Thus, the treating psychiatrists will be allowed to refer patients to appropriate mental health professionals for psychotherapy and electroconvulsive therapy if deemed clinically appropriate. However, any patients who receive depression-specific empirical psychotherapies (CBT and IPT) and electroconvulsive therapy will be considered to have deviated from the study protocol. The patient, however, will not be considered to have dropped out of the study at this phase and will receive protocol assessments. Although awareness of CBT has increased among Japanese mental health professionals following the approval of CBT as treatment for mood disorder by Japan's national health insurance scheme in 2010, the number of mental health facilities capable of providing CBT is still very limited. The current situation for IPT in Japan is similar.
Discontinuations
Discontinuation of intervention phase
If patients meet any of the following criteria, the treating psychiatrist will discontinue the study intervention. The patient will not be considered to have dropped out of the study but will be invited to enter the follow-up phase and will receive periodic assessments throughout the remainder of the study period.
-
The patient withdraws consent to receive the study intervention.
-
The treating psychiatrist judges that it is inappropriate to continue the study intervention due to, for example, the emergence of a severe psychotic or manic episode, serious and imminent suicidal ideation, or a severe medical condition.
-
The treating psychiatrist judges that it is difficult to continue the study intervention because of the emergence of adverse events or another appropriate reason that outweighs the benefit of receiving the study intervention.
-
The treating psychiatrist judges that it is more appropriate to receive inpatient psychiatric care.
Discontinuation of periodic assessments
If the patient withdraws consent to receive periodic study assessment, they will be considered to have dropped out and will not be contacted for periodic assessment in the future.
Outcome measures
The outcome measures are shown in table 2.
Schedule of the assessments
Primary outcome
The primary outcome is the change in the clinician-rated 17-item GRID-HAMD score at 16 weeks, which is when the intervention ends. The GRID-HAMD will be also administered at week 8 (intervention midpoint). Follow-up assessments will be administered at the 3-month (7 months after randomisation), 6-month (10 months after randomisation) and 12-month follow-up visits (16 months after randomisation). All assessors (psychiatrists and licensed clinical psychologists) have received extensive GRID-HAMD training and have achieved excellent inter-rater reliability (ICC=0.98). The GRID-HAMD will be administered by an assessor blind to treatment randomisation. Due to the nature of the intervention, neither the patients, the treating psychiatrists or the study therapists can be completely blinded to randomisation, but the two latter groups are strongly instructed not to disclose the randomisation status of the patient at periodic assessments. Further, the assessors will not be present during treatment administration.
Secondary outcomes
Clinical outcomes
-
Severity and change in scores of subjective depression symptoms as measured by the QIDS-SR16 and BDI-II.
-
Proportion of responders, defined as a 50% or greater reduction in scores for the 17-item and 21-item GRID-HAMD, BDI-II and QIDS-SR16 relative to baseline.
-
Proportion of patients who achieve remission, defined as a 17-item GRID-HAMD score ≤7,49 BDI-II score ≤1350 and QIDS-SR16 score ≤5.38
Safety outcomes
-
The proportion of patients who discontinue the study will be recorded. The patient will be asked at the site or by telephone for their reason for discontinuation, which will be ascertained by the treating psychiatrist.
-
Spontaneously reported adverse event (AEs) and serious adverse events (SAEs).
Health outcomes
-
Level and change in health-related quality of life as measured by the EQ-5D and SF-36.
Work performance outcomes
-
Self-reported sick leave hours (absenteeism), reduction in job performance (presenteeism), and the actual hours worked in the past 4 weeks as measured by the HPQ.
Economic evaluation
-
Quality of life (EQ-5D) and depression severity (GRID-HAMD, BDI-II and QIDS-SR16) will be used for estimating quality adjusted life years (QALYs) for cost-utility analyses.
Instruments
GRID-Hamilton Depression Rating Scale (GRID-HAMD)
The Hamilton Depression Rating Scale (HAMD) has been the gold standard assessment for observer-rated depression symptomatology for more than 50 years. The GRID-HAMD was developed to set standards for scoring and administering the original HAMD. Symptom severity is usually assessed for the 7 days before assessment. The GRID-HAMD has three components: the GRID scoring system based upon assessment of symptom intensity and symptom frequency, manual of scoring conventions, and a semi-structured interview guide based on the SIGH-D.51 The inter-rater reliability of the Japanese version of the GRID-HAMD total score is excellent.30
Beck Depression Inventory-Second Edition (BDI-II)
The BDI-II is a widely used self-report instrument to assess the severity of depressive symptoms and was developed by Beck and colleagues, with its first version published in 1961.52 The BDI-II is a 21-item questionnaire and each item is answered by circling a number between 0 and 3, with larger numbers indicating greater severity. Symptom severity for the BDI-II should be assessed for the previous 2 weeks to better coincide with DSM criteria. Good reliability and validity have been reported for the original36 as well as the Japanese version.50
16-Item QIDS-SR16
The QIDS-SR16 is an abbreviated self-report version of the clinician-rated 30-item Inventory of Depressive Symptomatology (IDS), designed to assess the severity of depressive symptoms, which was developed by John Rush and colleagues. The QIDS-SR16 assesses all the criterion symptom domains for diagnosing a DSM Major Depressive Episode. Symptom severity is usually assessed for the 7 days before assessment. Internal consistency is excellent with a Cronbach's α ranging from 0.81 to 0.94; validity is also very good with high correlation with HAMD38 as well as the Japanese version.39
36-Item Short-Form Health Survey (SF-36)
The SF-36 is a multi-purpose health survey with 36 items. It assesses the eight health domains of functional health (vitality, physical functioning, bodily pain, general health perceptions, physical role functioning, emotional role functioning, social role functioning, and mental health), level of well-being, physical and mental health summary measures, and a health utility index. Good validity has been reported for the original53 as well as the Japanese version.42
WHO Health and Work Performance Questionnaire (HPQ)
The HPQ is the most widely used self-report instrument designed to estimate the workplace costs of health problems in terms of reduced job performance, sickness absence, and work-related accidents and injuries. It assesses work hours, sick leave, occupational accidents, and self-rated productivity in the past 7 days and past 4 weeks. The validity of the HPQ absenteeism and presenteeism measures has been confirmed.54
European Quality of Life Questionnaire–5 dimensions (EQ-5D)
The EQ-5D is a generic, multidimensional, health-related, quality-of-life instrument that consists of a health status profile and a visual analog scale (VAS) to rate global health-related quality of life.40 The health status profile covers five health domains (mobility, self-care, usual activities, pain/discomfort, and mood) and the score of the five domains will be mapped to a single index value through an algorithm. The index value ranges between 0 and 1 with a higher score indicating a better health state perceived by the patient. The index value is used for calculating QALYs. The EQ-5D is the instrument preferred by the National Institute for Health and Care Excellence (formerly the National Institute for Health and Clinical Excellence) to measure health-related quality of life in adults.7
Sample size estimation
The sample size is calculated based on the primary outcome of depression symptoms as measured by the 17-item GRID-HAMD score at 16 weeks after randomisation. Our previous single group study on CBT with treatment as usual for acute major depression has shown that the 17-item GRID-HAMD score will drop from 24.3 (SD 7.4) to 10.0 (SD 5.0) at week 16.47 We expect a mean difference of 40% (4 points) in the 17-item GRID-HAMD total scores between the groups at endpoint and consider this to be a clinically meaningful difference. With a two-sided significance level of 5% and statistical power at 90% and allowing for 15% drop-out, the sample size was calculated to be 40 per arm, that is, 80 in total.
Statistical analyses
The primary analysis population in this study will be the intention-to-treat group, defined as all randomised patients. For the primary outcome, the least squares means and their 95% CIs will be estimated using analysis of covariance (with treatment group as a factor and baseline scores as a covariate) to compare the two groups, with a last-observation-carried-forward approach for missing values. To examine the robustness of the last-observation-carried-forward approach, a mixed-effects model for repeated measures that contains treatment group, week, and group-by-week interaction as factors with a compound symmetry covariance matrix among time points, and Kenward–Roger degrees of freedom adjustment will be performed with all the primary outcomes and also for continuous secondary outcomes. Categorical outcomes will be analysed using the χ2 test or Fisher's exact test. Summary statistics (means and SD) of patients’ characteristics will be calculated. When appropriate, the t test and Mann–Whitney U test will be used to compare baseline continuous outcomes (means). Time to all cause discontinuation will be summarised using Kaplan–Meier estimates and compared with the log-rank test. The significance level will be set at 0.05 (two-tailed). Statistical analyses will be performed with SAS V.9.3.
Data collection and management
To ensure accurate, complete, and reliable data, the following procedures will be followed: (1) standardised operational procedure material regarding data collection, encoding, and storage will be provided to the study sites; (2) a training session will be held to provide instruction on the protocol, the completion of electronic data capture (EDC), and study procedures for study psychiatrists, study therapists, and study coordinators; (3) periodic meetings will be held with study site personnel to discuss issues related to the conduct of the study; (4) the principal and co-principal investigator will be available for consultation and stay in contact with the study site personnel by mail, telephone, and/or fax; and (5) a data manager will review and evaluate EDC data, use standard computer edits to detect errors during data collection, and conduct a quality review of the database.
To ensure the safety of the participants in the study and to ensure accurate, complete, and reliable data, the study psychiatrist will keep records of paper instruments and clinical records in patient files as source documents for the study at the site. The principal investigator (YO), the co-principal investigator (AN), the study statistician (TA) and other steering committee members (MS, DF, TK) will be given access to the cleaned data sets.
EDC system
An EDC system will be used in this study. The site will maintain the original source for the data entered by the site into the EDC system. The electronic case report form data collected by the study psychiatrists, therapists or clinical research coordinators will be encoded and stored electronically in the database. Data will be managed by the data manager at the Keio Center for Clinical Research and will be stored electronically in the database.
Study monitoring
The data manager at the Keio Center for Clinical Research will conduct periodic inspections of the accumulating outcome data throughout the course of the study. The Data Safety Monitoring Committee (DSMC) may request additional evaluation or follow-up of patients who have clinically significant events.
Interim analyses
Interim analyses are planned to assess for safety and ineffectiveness when 50% of patients (n=40) have been randomised and have completed the 16-week post-randomisation assessment. The interim analysis will be performed by a member of the DSMC who is blind to the allocated treatment. The incidence of SAEs in the sample and the 17-item GRID-HAMD score after treatment (16 weeks) will be compared between groups to determine if the intervention is futile (ie, a 15% or less mean difference between the groups). The results of the interim analyses will be discussed with the principal investigator, who will decide whether to continue, stop, or modify the trial.
Premature termination of the study
The study will be terminated if the principal investigator, upon advice from the DSMC, judges it necessary for medical safety, for instance if a causal relationship between the study intervention and SAEs is established or there is a serious ethical violation according to the Ethics Guideline for Clinical Research (Ministry of Health, Labour and Welfare, revised in 2008).
Reporting of adverse events
All adverse events reported spontaneously by patients or observed by the treating psychiatrists will be recorded. When an adverse event occurs, the treating psychiatrist will take all necessary and appropriate measures to ensure the safety of the patient.
When an SAE occurs, the treating psychiatrist must take all necessary and appropriate measures to ensure the safety of the study patient and must provide appropriate treatment including hospital admission. Based on the Ethics Guideline for Clinical Research (Ministry of Health, Labour and Welfare, revised in 2008) an SAE is defined as ‘an adverse event that may lead to death or to enduring severe impairment depending on the patient's conditions and circumstances’ and will include: (1) death (all deaths regardless of causal relationship with the intervention or whose causal relationship with the intervention cannot be excluded, during the intervention phase or up to 30 days after the completion of the intervention); (2) a life-threatening event; (3) an event leading to enduring and severe impairment and dysfunction; and (4) hospitalisation (all hospitalisation regardless of causal relationship with the intervention or whose causal relationship with the intervention cannot be excluded, during the intervention phase or up to 30 days after the completion of the intervention).The treating psychiatrist must notify the principal investigator (YO) of the SAE immediately, and the principal investigator must also notify all collaborating investigators. The head investigator of the study site must report the SAE to its own ethics review committee and, if it concerns an unforeseeable SAE, must report it to the Japanese Ministry of Health, Labour and Welfare.
Ethical considerations and dissemination
Ethics approval of the study protocol was obtained from the Ethics Review Committee of Keio University School of Medicine (reference no. 20070070, 19-70-4) and the Ethics Review Committee of Sakuragaoka Memorial Hospital. The trial is registered in the UMIN Clinical Trials Registry: UMIN000001218.
Informed consent
The study psychiatrist is responsible for ensuring that the patient understands the potential risks and benefits of participating in the study, and should answer any questions the patient may have throughout the study and share in a timely manner any new information that may be relevant to the patient's willingness to continue his or her participation in the study.
An informed consent form will be used to explain the potential risks and benefits of study participation to the patient in simple terms before the patient is entered into the study and to document that the patient is satisfied with his or her understanding of the risks and benefits of participating in the study and desires to participate in the study. The study psychiatrist is responsible for ensuring that informed consent is given by each patient. The appropriate signatures and dates on the informed consent form must be obtained before administration of the intervention.
Ethics review
The principal investigator (YO) and the co-principal investigator (AN) must agree on the protocol and informed consent form before they are submitted to the ethics review committee and are used at sites. The protocol and informed consent form must comply with the Ethics Guideline for Clinical Research (Ministry of Health, Labour and Welfare, revised in 2008). The ethics review committee will review the protocol as required. When the protocol needs to be amended for a legitimate reason, such as safety concerns, the protocol will be revised and after the agreement of the principal investigator and the co-principal investigator, will be submitted to the ethics review committee for review.
Compensation and insurance for harmed patients
We cannot guarantee that unforeseen serious complications or health damage will not occur during or after completion of participation in this study. If such do occur, appropriate measures will be taken, the same as in cases of treatment for health damage in usual medical care. The medical expenses shall be borne by the patient, since the treatment will be supplied as a healthcare service provided under national health insurance, the same as for usual treatment. There will be no special financial compensation; however, any negligence on the part of the physician may be covered by the doctors’ liability insurance.
Conflict of interest
Research objectivity and commitment to academic integrity is of paramount importance and the basis for obtaining and maintaining public trust, so all investigators will comply with the site's policy on conflicts of interest in research and relevant conflict of interest guidelines.
Dissemination
The results of the study will be disseminated at several research conferences and as published articles in peer-reviewed journals. The study will be implemented and reported in line with the CONSORT statement.
Discussion
The Effectiveness of Cognitive Behavioural Therapy Augmentation in Major Depression Treatment study (ECAM study) aims to provide new evidence concerning the administration of CBT for major depression as a second-line therapy for patients who have failed to respond to pharmacotherapy in psychiatric care settings. The design of the study is expected to detect a meaningful difference in clinical effectiveness outcomes. The ECAM study differs from previous studies in that the study design uses a standardised psychiatric interview to assess depression symptomatology by blind raters, recruits patients from secondary and tertiary psychiatric care facilities who tend to have more severe and more difficult-to-treat depression, and evaluates the long-term effects of CBT for up to 12 months.
The challenge and limitation of this study is that we cannot examine the efficacy of CBT itself because we did not choose an attention placebo, such as relaxation, as control. Our aim is to conduct a study to examine the effectiveness of augmenting usual clinical care with CBT rather the efficacy of CBT itself. We are also aware that the participating sites in this study are, clinically speaking, experienced in the treatment of patients with depression. Thus, the results might not be the generalisable to other settings. Nevertheless, this is the first randomised controlled study to assess the effectiveness of CBT for TRD in Japan. The results of the current study will hopefully improve the evidence-based treatment of patients who have residual symptoms of depression despite adequate pharmacotherapy.
Current study status
The ECAM study began recruiting patients in September 2008 and closed recruitment in August 2013. Data collection will be completed in December 2014.
Acknowledgments
The authors would like to thank our study sites for their support. Clinical ratings will be completed by the clinical evaluation staff at the Sakuragaoka Memorial Hospital and the Department of Neuropsychiatry, Keio University School of Medicine. Thanks are also due to Dr Kimio Yoshimura, a member of Data Safety Monitoring Committee. We appreciate the informatics support provided by Yoko Ito, Kayoko Kikuchi, and Naoki Tomotsugu of the Project Management Office at the Keio Center for Clinical Research during the construction and data management of the electronic data capture system.
References
Footnotes
-
Contributors AN and MS conceived and designed the study. AN drafted the protocol of the study, and organised and supervised study implementation. DF and TK refined the study protocol and study implementation. TA and YS provided methodological and statistical expertise. TA and AN conducted the statistical analyses. YO provided CBT expertise and supervised the therapists. YO drafted the grant proposal and was responsible for study implementation. AN was responsible for study management, staff training, and supervision. DM managed day-to-day study responsibilities, including monitoring recruitment, collecting data, and liaising with recruitment sites. SI and MM are the directors of the two sites and provided clinical expertise and on-site management of the study. All authors critically reviewed and approved the final version of the manuscript.
-
Funding This study is supported by Health Labour Sciences Research Grants (H22-Seishin-Ippan-005, H25-Seishin-Ippan-002) from the Japanese Ministry of Health, Labour and Welfare. The funding source has no role in study design; in data collection, analysis and interpretation; in the writing of the report; or in the decision to submit the paper for publication.
-
Competing interests AN, MS, DF, TK and YO developed and wrote the Japanese CBT manual for depression, and are involved in the National CBT Training and Supervision Project funded by the Japanese Ministry of Health, Labour and Welfare. YO is president of the Japanese Association for Cognitive Therapy. None other authors have any conflicts of interest to declare.
-
Ethics approval The Ethics Review Committee of Keio University School of Medicine and the Ethics Review Committee of Sakuragaoka Memorial Hospital approved this study.
-
Provenance and peer review Not commissioned; externally peer reviewed.