Article Text

Abstract
Introduction Effective cardiopulmonary resuscitation with appropriate airway management improves outcomes following out-of-hospital cardiac arrest (OHCA). Historically, tracheal intubation has been accepted as the optimal form of OHCA airway management in the UK. The Joint Royal Colleges Ambulance Liaison Committee recently concluded that newer supraglottic airway devices (SADs) are safe and effective devices for hospital procedures and that their use in OHCA should be investigated. This study will address an identified gap in current knowledge by assessing whether it is feasible to use a cluster randomised design to compare SADs with current practice, and also to each other, during OHCA.
Methods and analysis The primary objective of this study is to assess the feasibility of a cluster randomised trial to compare the ventilation success of two newer SADs: the i-gel and the laryngeal mask airway supreme to usual practice during the initial airway management of OHCA. The secondary objectives are to collect data on ventilation success, further airway interventions required, loss of a previously established airway during transport, airway management on arrival at hospital (or termination of the resuscitation attempt), initial resuscitation success, survival to intensive care admission, survival to hospital discharge and patient outcome at 3 months. Ambulance paramedics will be randomly allocated to one of the three methods of airway management. Adults in medical OHCA attended by a trial paramedic will be eligible for the study.
Ethics and dissemination Approval for the study has been obtained from a National Health Service Research Ethics Committee with authority to review proposals for trials of a medical device in incapacitated adults. The results will be made publicly available on an open access website, and we will publish the findings in appropriate journals and present them at national and international conferences relevant to the subject field.
Trial registration ISRCTN: 18528625.
- Out of hospital cardiac arrest (OHCA)
- cardiopulmonary resuscitation (CPR)
- airway management
- tracheal intubation
- supraglottic airway devices (SADs)
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/3.0/ and http://creativecommons.org/licenses/by-nc/3.0/legalcode
Statistics from Altmetric.com
- Out of hospital cardiac arrest (OHCA)
- cardiopulmonary resuscitation (CPR)
- airway management
- tracheal intubation
- supraglottic airway devices (SADs)
Article summary
Article focus
-
Cardiopulmonary resuscitation (CPR) is fundamental to survival and a subsequent quality of life following out-of-hospital cardiac arrest (OHCA). A core component of CPR is effective airway management.
-
Historically, tracheal intubation has been used by paramedics to manage the airway during resuscitation for OHCA, but it has recently been proposed that newer supraglottic airway devices (SADs) may achieve better patient outcomes.
-
We are conducting the feasibility work for a large-scale randomised trial to compare current airway management with SADs in OHCA.
Key messages
-
Conducting prehospital research is practically and ethically challenging.
-
We have developed a model of consent that has gained NHS Research Ethics Committee and patient group approval.
-
It is important to consider a variety of outcomes, including health-related quality of life and cost-effectiveness.
Strengths and limitations of this study
-
The protocol has a clear focus on the engagement of paramedics and the ambulance service; this is fundamental to the success of prehospital trials.
-
This feasibility study has been designed for one ambulance service; variation between services and issues of scale will need to be considered in the full trial.
Introduction
Out-of-hospital cardiac arrest (OHCA) is a leading cause of death affecting nearly 300 000 people each year in Europe.1 Effective cardiopulmonary resuscitation (CPR) improves health-related quality of life and survival outcomes,2–4 but nevertheless most patients do not survive. Appropriate management of the patient's airway is regarded as a fundamental and essential component of CPR.5 ,6
Historically, intubation with a tracheal tube was viewed as the best form of prehospital airway management in cardiac arrest.6 This was based on extrapolation from in-hospital practice and has never been well tested in prehospital care. It has become apparent that significant complications can occur as a result of prehospital tracheal intubation.7 These include unrecognised oesophageal intubation or unrecognised dislodgement of the tracheal tube, which can be fatal.8 ,9 Intubation attempts may cause interruptions in chest compressions, compromised oxygenation and delays in accessing definitive care.10 This and other evidence has recently led the Airway Working Group of the Joint Royal Colleges Ambulance Liaison Committee (JRCALC) to conclude that current practice may not be beneficial to patients, and the use of newer supraglottic airway devices (SADs) should be investigated as a priority.11
The laryngeal mask airway supreme (LMAS) is a single-use SAD with an enhanced cuff designed to improve ventilation success and reduce the risk of aspiration.12 ,13 The characteristics of the LMAS make it a candidate for the optimal prehospital resuscitation airway. However, the same can also be said of the recently introduced i-gel. The i-gel is an SAD which uses a gel to provide an airway seal, rather than an inflatable cuff. Its insertion time was found to be faster than that of an older SAD in one manikin study,14 and it was rated as the easiest of eight SADs in another manikin study.15
This trial “Randomised comparison of the effectiveness of the laryngeal mask airway supreme, i-gel and current practice in the initial airway management of pre-hospital cardiac arrest: a feasibility study (REVIVE-Airways),” is a cluster randomised study to assess the feasibility of conducting a trial of usual paramedic practice versus the i-gel versus the LMAS on ventilation success during airway management in OHCA.
Methods and analysis
Trial management
National Research Ethics Service approval for this trial has been obtained from the Cambridge Central Research Ethics Committee and the trial is registered on the International Standard Randomised Controlled Trial Registry (ISRCTN: 18528625). The trial is funded by the National Institute for Health Research (NIHR) Research for Patient Benefit Programme (RFPB) and the Sponsor organisation is the University Hospital Bristol NHS Foundation Trust (UHBristol). REVIVE-Airways is a collaboration between UHBristol, Great Western Ambulance Service NHS Trust (GWAS), Royal United Hospital Bath NHS Trust (RUH) and the University of the West of England, Bristol. Research will be conducted within the Medical Research Council Good Clinical Practice Guidelines and the standard operating procedures of the sponsor organisation. The contribution of the manufacturers of the i-gel (Intrasurgical) and the LMAS (Intervent) will be limited to confirming that the training of paramedics in the use of the devices conforms to their recommended guidelines. These manufacturers will have no role in supplying devices or the design, conduct, analysis or reporting of the trial.
Design
This is a feasibility study using a cluster-randomised design to compare the effectiveness of the early use of the i-gel and LMAS with current paramedic practice during resuscitation for OHCA. Cluster randomisation has been adopted because an individually randomised design would have a significant danger of a high level of contamination among the arms. In an individually randomised design, all paramedics taking part in the trial would have to carry all devices, and there would be a strong possibility that they would use their preferred method of airway management. In addition, individual patient randomisation at the point that resuscitation started for OHCA would be both logistically and ethically challenging.
Objectives
The primary objective of this study is to assess the feasibility of a cluster randomised trial to compare the insertion and ventilation success of the i-gel versus LMAS versus current practice during the initial management of prehospital cardiac arrest. The secondary objectives are to describe the ventilation success of the i-gel versus LMAS versus current practice during the initial management of prehospital cardiac arrest, to estimate the intraclass correlation coefficient for paramedic clusters to inform subsequent calculations of sample size and to describe further airway interventions required, loss of a previously established airway during patient transport, airway management at the time of hospital arrival (or termination of the resuscitation attempt), initial resuscitation success, survival to intensive care admission and survival to hospital discharge.
Outcome measures
The primary outcome measure is ventilation success, defined as visible chest movement with each ventilation and audible air entry in both axillae on stethoscope auscultation. Secondary outcomes are: insertion success; regurgitation and aspiration; further airway interventions undertaken; loss of a patent airway during transport; airway management at hospital arrival or when resuscitation terminated; survival of event (sustained return of spontaneous circulation (ROSC) to arrival at hospital); organ dysfunction (Acute Physiology and Chronic Health Evaluation) score, intensive care length of stay; hospital length of stay; survival to hospital discharge, neurological status (Cerebral Performance Category (CPC)) and health-related quality of life (EQ-5D) at discharge. Further follow-up data will be collected from those discharged with neurologically intact survival to 3 months (survival with CPC score 1 or 2): health-related quality of life (SF36 and EQ-5D); anxiety and depression (Depression Anxiety and Stress Scale (DASS)); cognition (Mini-Mental State Examination (MMSE)) and neuropsychological outcome (CANTAB). Staff feedback: a short postal survey will be administered to all recruited paramedics who attend two or more cardiac arrests during the study period.
Eligibility
Paramedics working within GWAS who consent to participate in the study will be randomly allocated to one of the three study arms: i-gel, LMAS or control (usual airway practice). Randomisation will be stratified by years of paramedic experience (greater than or less than 4 years full-time operational experience) and urban/rural location of the base ambulance station. Paramedics will undergo appropriate training for their study arm, be issued with a personal supply of the relevant device (i-gel and LMAS arms) and then be required to account for each use.
Patients will be eligible if they are:
-
In OHCA;
-
Attended by a paramedic participating in the trial;
-
Attempted resuscitation is appropriate according to JRCALC guidelines;
-
Known or believed to be 18 years or older.
Exclusion criteria:
-
Patient is less than 18 years old;
-
Cardiac arrest caused by trauma;
-
Estimated weight is less than 50 kg;
-
Mouth opening is less than 2 cm.
Treatment allocation of each patient will be determined by the first enrolled paramedic who arrives on the scene: if the paramedic is randomised to the i-gel, the patient will be included in the i-gel arm; if the paramedic is randomised to the LMAS, the patient will be included in the LMAS arm and if the paramedic is randomised to usual practice, the patient will be in the control arm. If the first response to arrive on the scene is a community responder or other ambulance response not participating in the trial, then the patient will be included and their allocation will be determined by the first trial paramedic to arrive on the scene, providing that continued resuscitation is indicated. Figure 1 shows the flow of patients through REVIVE-Airways.

Eligibility flow chart.
Sample size considerations
Cluster size
Data collection will take place over 1 year. There are around 450 eligible paramedics working within GWAS, and audit data show that approximately 240 cardiac arrests occur each month throughout the service, of which approximately half are eligible for inclusion in this study. Assuming that one-third of eligible paramedics enrol, 150 will be available for randomisation (50 per group) and these paramedics will attend approximately 480 eligible cardiac arrests during the 12-month study period.
Assuming 80% complete data collection, data will be available from 384 cardiac arrests, or 128 patients per group which should yield a mean number of events per practitioner of 2.6. If these events are distributed randomly among 50 paramedics, following a Poisson distribution, it is estimated that the number of paramedics who attend two or more events in 12 months will be 36 in each group.
Sample size required
In this feasibility study, a formal calculation of power has not been carried out, but an analysis of the expected number of paramedics who would be able to attend two or more eligible events in a 12-month period has been carried out, estimating the events to be occurring randomly throughout the sample and following a Poisson distribution. This reveals that 7% of paramedics would not be expected to attend any suitable patients in 12 months, and 10% would attend only one event. In order to achieve at least 30 paramedics in each group who have attended at least two events, and to allow for some paramedics to leave GWAS during the study, 50 will be recruited to each group.
Ethical considerations
Approval has been obtained from a National Health Service (NHS) Research Ethics Committee with authority to review proposals for trials of a medical device in incapacitated adults.
GWAS paramedics will be invited to participate in the study through a process of informed consent. Recruitment of paramedics raises no particular ethical issues since they are NHS clinicians who are able to consider the study over a period of time.
The enrolment of patients in cardiac arrest without consent is ethically challenging and requires special consideration. Conducting research in emergency situations where a patient lacks capacity is regulated by the Mental Capacity Act16 for England and Wales.
The occurrence of a cardiac arrest out of hospital is unpredictable, and there is no alternative patient group in which the research question could be meaningfully addressed. Within seconds of cardiac arrest, a person becomes unconscious and is thus incapacitated. As a result, it is impossible to obtain prospective consent from the patient. Treatment (in the form of CPR) must be started immediately in an attempt to save the person's life. In this setting, it is also impractical to consult a carer or independent-registered medical practitioner without placing the potential participant at risk of harm from delaying treatment. Therefore, it is impractical to seek any form of consent before enrolment and intervention. If the patient does not survive to Intensive Care Unit (ITU) discharge, consent will not be sought retrospectively; the trial intervention has been completed by the time the patient reaches hospital, and in these circumstances consent cannot be meaningfully given or withheld subsequently. Furthermore, to approach the relatives of a patient who has recently died following OHCA is likely to cause avoidable distress without gain. Recent changes in research consent in the UK support this model, and a precedent has been established by a large trial of an automated external compression device during prehospital cardiac arrest funded by the Health Technology Assessment Programme of the NIHR.17 This approach to consent has also been specifically debated and endorsed by our patient forum, as well as being approved by the trial funding body and research ethics committee. Therefore, we will only seek consent (to collect follow-up data) from those patients who survive to ITU discharge.
Approaching survivors for consent
The nature of the condition means that the majority (>90%) of people in the study will not survive. Of those patients admitted to hospital alive, the majority (approximately 90%) will be comatose and admitted to an intensive care unit (and thus remain incapacitated for several days at least).
The timing of the approach is important and needs to balance the need to inform at an early opportunity while determining accurately which patients have died, and which are potentially able to give consent. All enrolled patients that survive to hospital admission will be followed up by the trial coordinator, who will consult with hospital staff to determine the optimal time to approach the patient and/or their family to seek consent for further follow-up and data collection. Consent will usually be obtained upon discharge from the ITU. An information sheet will be provided and written consent obtained. Once written consent has been obtained, the patient's general practitioner will be sent an information letter detailing the study.
Protection against bias
Cluster design
One of the major potential sources of bias in cluster randomised trials is inclusion of different patients in the arms of the trial. This can arise where a large proportion of potentially eligible patients are not included in the trial, and the probability of inclusion is related to the intervention. In this trial, we aim to identify and include close to 100% of the eligible patients, using a combination of methods for identifying eligible patients, including direct notifications by paramedics and a review of routine ambulance service data.
Threshold for resuscitation
The criteria that are used to determine whether a resuscitation attempt is appropriate, and hence whether the patient is eligible, are as objective as possible. The JRCALC Recognition of Life Extinct (ROLE) criteria are used by GWAS to determine when a resuscitation attempt is inappropriate, and this will continue in the trial. However, there is a possibility that bias could be introduced by different thresholds for resuscitation between the three trial arms, as paramedics delivering the interventions will not be blinded. We will instigate a programme of regular monitoring by analysing the characteristics of patients recruited to the three arms and cardiac arrests where no resuscitation attempt was made, and the proportion of cardiac arrests recruited, to detect any imbalances that may be caused by different thresholds for resuscitation.
Blinding
Owing to the nature of the intervention, ambulance paramedics cannot be blinded and will be aware of treatment allocations. Control room personnel will be blinded to the allocation of paramedics and follow established protocols when allocating resources to a possible cardiac arrest. This will ensure that there is no bias in despatch. Patients themselves will be unaware of their treatment allocation at the time of the intervention, and this is likely to be maintained throughout the trial. We will seek to ensure blinding of outcome assessment as far as possible. Mortality is an objective outcome whose assessment will not be influenced by knowledge of the treatment allocation. Research staff assessing outcomes at the 3 month follow-up will be blinded to the treatment group.
Trial interventions
LMAS arm
Patients in this arm will receive resuscitation according to the Resuscitation Council (UK) and JRCALC Advanced Life Support Guidelines, with the exception that the LMAS device will be used to manage the airway. All standard advanced life support interventions will be provided including drug administration, defibrillation and chest compressions as required.
Chest compressions will be started or continued according to standard resuscitation protocols. The LMAS will be inserted at the first opportunity without interrupting CPR, and assisted ventilation will be provided at a rate of approximately 10 breaths a minute while compressions are ongoing. If ventilation is deemed inadequate (no chest rise observed) while compressions are ongoing, ventilations will be provided at a rate of 2 : 30 compressions while pausing compressions. If the patient has vomited before LMAS insertion, the airway will be suctioned first. If insertion or ventilation is unsuccessful, the clinician will use any alternative airway and ventilation technique that they deem to be in the best interests of the patient according to their personal skills and the clinical situation.
i-Gel arm
This arm will receive resuscitation according to the Resuscitation Council (UK) and JRCALC Advanced Life Support Guidelines, with the exception that the i-gel device will be used to manage the airway. All standard advanced life support interventions will be provided including drug administration, defibrillation and chest compressions as required. Trial intervention will be identical as described above for the LMAS arm, except that an i-gel will be used in place of an LMAS.
Control arm
This arm will receive resuscitation according to the Resuscitation Council (UK) and JRCALC Advanced Life Support Guidelines. All standard advanced life support interventions will be provided including drug administration, defibrillation and chest compressions as required.
The practitioner will employ the airway management methods and ventilation technique that they deem to be in the best interests of the patient according to their personal skills and the clinical situation—this may include tracheal intubation or the use of a disposable AMBU LMA issued by GWAS.
Postresuscitation care (all arms)
The care that a patient receives in hospital following ROHC has a significant influence on final outcome. There is no reason to suppose that patients treated in the SAD arms as opposed to the usual practice arm would receive any different treatment in hospital, but we will document this during patient follow-up to ensure that it is the case.
Data collection
Incidents of cardiac arrest occurring within the region covered by GWAS will be identified on a daily basis by searching data from Computer Aided Despatch (CAD) reports. Those arrests attended by a trial paramedic will be identified. In addition, trial paramedics will be instructed to report each arrest they attend to the study coordinator and complete a Case Report Form (CRF). In the event that an eligible arrest has been identified as being attended by a trial paramedic, but not reported by them, the trial coordinator will contact the paramedic to request that a CRF be completed retrospectively. Data from the routinely collected GWAS cardiac arrest registry will also be searched to identify any eligible arrests that occurred but were not coded as arrests on the CAD report; if a trial paramedic attended a cardiac arrest that is detected in this way, they will again be contacted and asked to complete a CRF retrospectively. In this way, we hope to identify and collect data from every eligible cardiac arrest that occurs during the study period.
Training
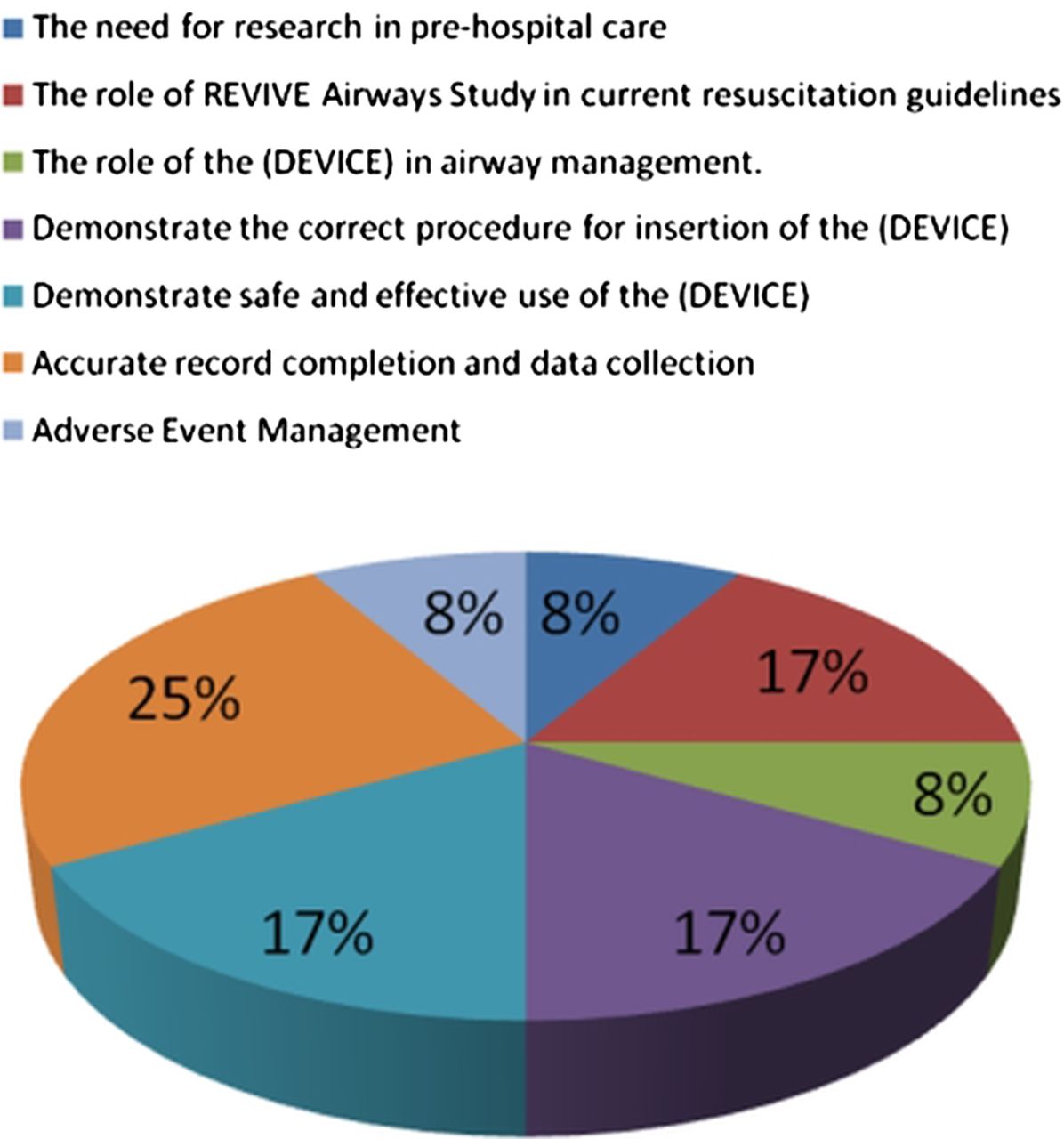
All paramedics recruited to the study will undergo a structured training session on the purpose and protocol of the trial and familiarisation with the CRF. The session will review current resuscitation guidelines and involve a practical session using a manikin to practise resuscitation techniques and protocols, particularly effective airway management. For paramedics randomised to the i-gel or LMAS arms, specific training on the use of their allocated device will be provided in accordance with the manufacturer's guidelines; representatives from the manufacturers will attend a pilot training session to endorse the content of the training and mechanism of delivery, but will not be engaged in any other way in the design, funding, conduct or reporting of the study. Competency will confirmed by the administration of a brief, standardised verbal and practical assessment. Regular ongoing training in SADs and the trial procedures will be conducted as required, during visits that will be made to each station. The trial coordinator will maintain records of all personnel who have been trained in the use of SADs. Figures 2 and 3 describe the content of the training and the time allocation to the different elements.
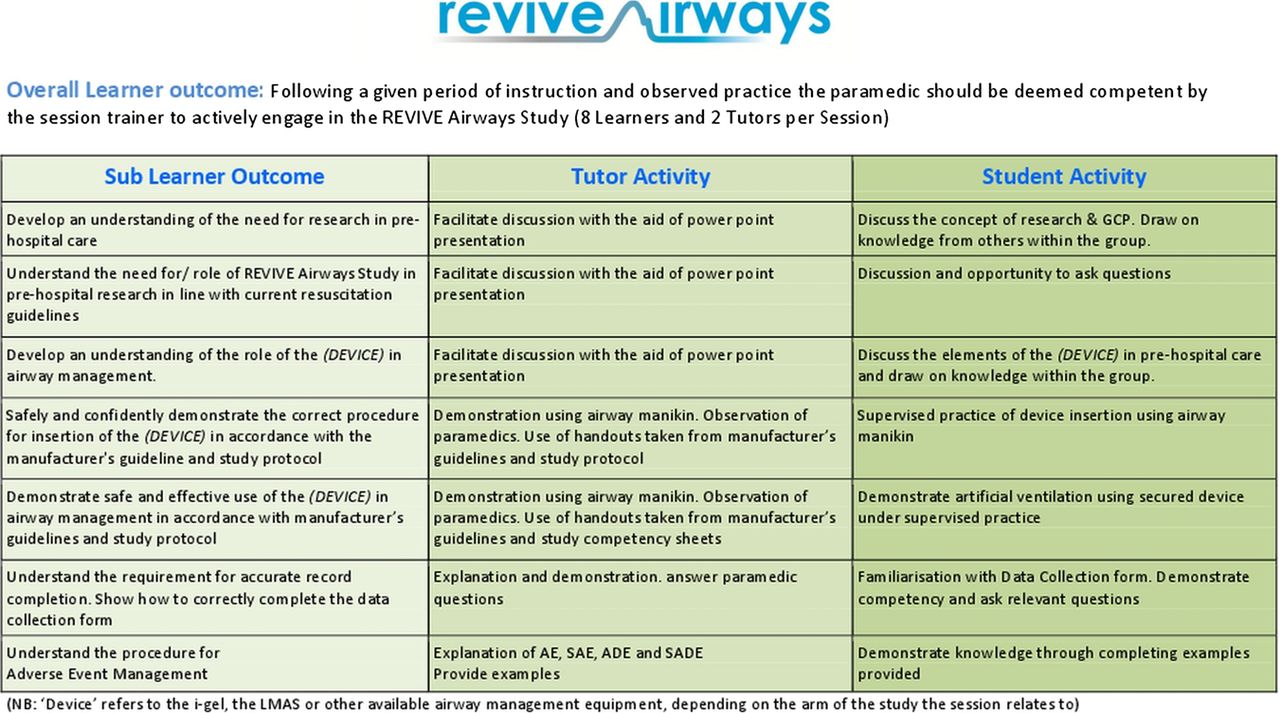
Paramedic training course content.
{kind=link}
{kind=link}
{kind=link}
Time allocation for paramedic training (percentage of total session).
Patient follow-up
Patients who survive to hospital discharge and have consented to follow-up will be approached 3 months after their cardiac arrest. They will receive a home visit from a trial researcher and complete the SF36, EQ-5D and DASS. The MMSE and selected tests from the CANTAB will be administered by the researcher.
Serious adverse event management
Serious Adverse Events (SAEs) and Serious Adverse Device Events (SADEs) will be reported in accordance with the Sponsor's Research Related Adverse Event Reporting Policy.
All of the patients in this trial will be in an immediately life-threatening situation, many of whom will not survive, and all of those that do will be hospitalised. SAEs and SADEs will be reported if they fulfil the criteria for seriousness, they are potentially related to trial participation and they are unexpected (ie, the event is not an expected occurrence for patients who have had a cardiac arrest).
End of the trial
The trial will end when the 3 month follow-up of the last patient has been completed. The trial will be stopped prematurely if: mandated by the Ethics Committee; the TSC decides that recruitment should cease following recommendations from the DMC; funding for the trial ceases.
Data analysis
Feasibility outcomes analysis
The recruitment rates of participating paramedics will be calculated as a percentage and 95% confidence limits ascribed using the exact binomial method. This will be carried out for those who were originally recruited compared to those eligible for recruitment, and also for those who were still part of the study at the end of the study period (had not changed their role or moved job and were still co-operating with the study), compared with those who were eligible at the start of the study.
All cardiac arrests that are eligible for the study will be identified both through the attending paramedics and through routine GWAS data monitoring systems. The percentage of these events reported by the attending paramedics will be compared with those considered eligible through routinely collected sources. The percentage of eligible arrests included by the paramedics for each of the randomised groups will be compared using Fisher's exact test.
The intraclass correlation of the binary outcome variable survival will be calculated using a random effects model in which the data are grouped by paramedics. This is required for the planning of the full study sample size. This will produce both the estimate of the intraclass correlation coefficient and the 95% CI of the estimate.
Full trial analysis
Should a full trial follow this feasibility study, it will be assessed using the outcome of the cardiac arrest, primarily in terms of survival rates and secondarily in terms of the overall quality of life following the arrest.
Planned dissemination
A dissemination strategy will be implemented that includes electronic dissemination of the results to the ambulance service and the staff who participated. The results will be made publicly available on an open access website, and we will publish the findings in appropriate journals and present them at national and international conferences relevant to the subject field. The feasibility data are important to inform further research in this area, while the clinical data are significant since this is the first detailed description of usual airway management during OHCA in the UK. We will therefore publish the clinical data in addition to the feasibility findings, while emphasising that the study was neither designed nor powered to demonstrate a clinically significant difference between the trial arms, and a further full-scale trial is therefore needed to definitively identify the best approach to initial airway management in OHCA.
Conclusion
Effective airway management is an integral component of CPR, and recent UK guidelines have called for newer airway devices to be investigated as a priority. The optimal method of airway management during OHCA has yet to be determined; this is in part due to the practical and ethical challenges associated with prehospital research. REVIVE-Airways is a feasibility study that has been designed to address these challenges and pave the way for a full-scale trial to evaluate the clinical effectiveness and cost-effectiveness of alternative strategies for airway management during OHCA.
References
Footnotes
-
Trial Steering Committee Membership Professor Simon Gates (Chair), Professor Jonathan Benger (Trial Chief Investigator), Dr Charles Deakin, Ms Anne Keat, Mr Ben Morgan, Ms Debbie McPhee, Professor Barney Reeves and Dr Jasmeet Soar.
-
Data Monitoring Committee Dr Gordon Taylor (Chair), Professor Gavin Perkins, Professor Helen Snooks and Professor Tom Quinn.
-
Contributors JRB drafted the research protocol, which was further developed by DC, RG, JN, SR and MT. MR developed the paramedic training course. SV drafted the manuscript for publication, which was further edited and approved by all authors. JRB is the study chief investigator and acts as the guarantor.
-
Funding This article presents independent research funded by the National Institute for Health Research (NIHR) under its Research for Patient Benefit (RfPB) Programme (Grant Reference Number PB-PG-0110-20288). The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
-
Competing interests None.
-
Ethics approval East of England—Cambridge Central.
-
Provenance and peer review Not commissioned; internally peer reviewed.