Article Text

Abstract
Objectives To assess the safety of an AS03-adjuvanted split virion H1N1 (2009) vaccine (Pandemrix) in persons vaccinated during the national pandemic influenza vaccination campaign in the UK.
Design Prospective, cohort, observational, postauthorisation safety study.
Setting 87 general practices forming part of the Medical Research Council General Practice Research Framework and widely distributed throughout England.
Participants A cohort of 9143 individuals aged 7 months to 97 years who received at least one dose of the AS03-adjuvanted H1N1 pandemic vaccine during the national pandemic influenza vaccination campaign in the UK was enrolled. 94% completed the 6-month follow-up. Exclusion criteria were previous vaccination with other H1N1 pandemic vaccine and any child in care.
Primary and secondary outcome measures Medically attended adverse events (MAEs) occurring within 31 days after any dose, serious adverse events (SAEs) and adverse events of special interest (AESIs) following vaccination were collected for all participants. Solicited adverse events (AEs) were assessed in a subset of participants.
Results MAEs were reported in 1219 participants and SAEs in 113 participants during the 31-day postvaccination period. The most frequently reported MAEs and SAEs were consistent with events expected to be reported during the winter season in this population: lower respiratory tract infections, asthma and pneumonia. The most commonly reported solicited AEs were irritability in young children aged <5 years (61.8%), muscle aches in children aged 5–17 years (61.9%) and adults (46.9%). 18 AESIs, experienced by 14 patients, met the criteria to be considered for the observed-to-expected analyses. AESIs above the expected number were neuritis (1 case within 31 days) and convulsions (8 cases within 181 days). There were 41 deaths during the 181-day period after vaccination, fewer than expected.
Conclusions Results indicate that the AS03-adjuvanted H1N1 pandemic vaccine showed a clinically acceptable reactogenicity and safety profile in all age and risk groups studied.
Trial registration ClinicalTrials.gov, NCT00996853.
- Epidemiology
- Immunology
- Infectious Diseases
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
Article summary
Article focus
-
The outbreak of the H1N1 (2009) influenza pandemic led to the vaccination of high-risk groups with novel pandemic vaccines targeting the A/California/7/2009 (H1N1)v-like strain. Limited data about the clinical safety of these novel vaccines were available.
-
In this paper, we report the results of a postauthorisation safety study designed as a pharmacovigilance activity to evaluate safety endpoints related to the H1N1 pandemic vaccination.
Key messages
-
The most frequently reported medically attended events and serious adverse events were consistent with events expected to be reported during the winter season.
-
The observed number of adverse events of special interest—Bell's palsy, Guillain-Barré syndrome and demyelination—were below the expected number.
-
The AS03-adjuvanted H1N1 (2009) vaccine was generally well tolerated in the age and risk groups studied, with clinically acceptable reactogenicity and safety profiles.
Strengths and limitations of this study
-
General practices, the primary point of contact for persons in the UK to access the National Health Service, were able to provide an extensive overview of the safety profile of the AS03-adjuvanted H1N1 pandemic influenza vaccine.
-
Sample size was not estimated for each risk group (immunocompromised, at-risk or healthy participants). Thus, it is difficult to ascertain whether the analysis reported here was sufficiently powered to adequately assess safety outcomes in the general UK population.
Introduction
Following the identification of several patients with swine-origin influenza who underwent human-to-human transmission,1–3 a Pandemic Alert announcement was issued by the World Health Organization (WHO). The lack of similarity of the pandemic virus strain to the current seasonal circulating influenza virus resulted in large-scale vaccination programmes, particularly in high-risk groups.4 ,5
In response, two pandemic vaccines were manufactured by GlaxoSmithKline Vaccines, including Pandemrix. This split-virion vaccine against the A/California/7/2009 H1N1 strain was adjuvanted with an α-tocopherol oil-in-water emulsion-based Adjuvant System containing qualene (AS03)6 ,7 and was produced in GlaxoSmithKline Vaccines’ Dresden (Germany) facility. The development of this vaccine was based on the experience acquired with H5N1 ‘mock-up’ vaccines.7–9 These H5N1 vaccines were highly immunogenic and had clinically acceptable safety profiles in children aged ≥6 months and adults.7–9
In response to this lack of available safety data, the European Medicines Agency (EMA) provided recommendations on pharmacovigilance activities that should be undertaken during the pre-pandemic and pandemic periods. During the 2009 pandemic influenza outbreak, the EMA recommended that vaccine manufacturers actively liaise with public health and regulatory authorities to explore the possibility of an association between A/H1N1 vaccines and severe adverse events.10 In the UK, a national immunisation programme against pandemic influenza was initiated in October 2009 by the UK Department of Health.11 ,12 Priority for vaccination was given to persons who were aged between 6 months and 65 years in the current seasonal influenza clinical risk groups: persons with chronic respiratory disease and asthma; chronic heart, renal, liver or neurological disease; diabetes or immunosuppression.11 ,13 The current UK study was suggested by the Medicines and Healthcare products Regulatory Agency (MHRA) and was implemented as a commitment to the authorities based on the recommendations of the EMA.
This study was a postauthorisation safety study (PASS) designed as a pharmacovigilance activity in addition to analysing signal detection from spontaneous adverse events (AEs) reporting. Data were provided promptly and periodically to the authorities after the study start. We have previously reported a preliminary analysis based on the cohort of women known to be pregnant at the time of vaccination in this study,14 and so pregnancy outcomes are not included in this report. Here, we discuss the other safety endpoints related to the H1N1 pandemic vaccination evaluated in all participants of this study.
Methods
Study design
This was a prospective, observational, multicentre, single cohort study of persons vaccinated with the H1N1 (2009) pandemic influenza vaccine (Pandemrix, GlaxoSmithKline Vaccines) in the UK. The study vaccine was produced in GlaxoSmithKline Vaccines Dresden, Germany. According to recommendations from the Committee for Medicinal Products for Human Use (CHMP) of the EMA,10 solicited AEs were planned to be assessed in a subset of 600 participants. The study was sponsored by GlaxoSmithKline as part of the AS03 adjuvanted H1N1 (2009) vaccine Risk Management Plan.
This study was conducted through general practices which were largely distributed throughout England and which were part of the Medical Research Council (MRC) General Practice Research Framework (GPRF). The vaccine was administered at the general practice according to the local pandemic influenza programme. Individuals were invited to participate in the study within 24 h after vaccination. General practices collected background information (such as demographics, relevant medical history), data on medication and vaccinations administered during the study, reactogenicity data via patient self-completed diary cards and safety data related to the study endpoints. Participants were contacted by the general practice or other delegated party at specific time points (24–96 h after any dose, 28–42 days after the last dose, 180–210 days after the last dose) to ensure that all clinical data pertaining to AEs were reported. The duration of the study was 7–8 months per participant; the first participant was enrolled on 31 October 2009 and the last participant was enrolled on 15 December 2009.
This study was conducted in accordance with good clinical practice (GCP) and all applicable regulatory requirements, including the Declaration of Helsinki. The study protocol and informed consent forms were reviewed and approved by a national Independent Ethics Committee. This study is registered at ClinicalTrials.gov (NCT00996853). A summary of the study protocol is available at http://www.gsk-clinicalstudyregister.com (Study ID 113585).
Study objectives
The primary objective of this study was to estimate the incidence of medically attended adverse events (MAEs) in all enrolled vaccinated participants within 31 days after vaccination. The secondary objectives were to assess vaccine reactogenicity within 7 days after vaccination, and to estimate the incidence of serious adverse events (SAEs) and adverse events of special interest (AESIs) in different age groups following an active surveillance of all enrolled vaccinated participants within 6 months after vaccination. An AESI was an event considered by the CHMP as worthy of closer follow-up as described in their recommendations for the Pharmacovigilance Plan following the administration of H1N1 pandemic vaccines. It included the following specific events for close monitoring: anaphylactic reaction, Bell's palsy, convulsion, demyelinating disorders, non-infectious encephalitis, Guillain-Barré syndrome (GBS), neuritis, vasculitis and vaccination failure.10
Study participants
Participants were included in the national H1N1 swine flu vaccination programme in the UK. Eligible participants included male and female persons vaccinated with at least one dose of H1N1 (2009) pandemic influenza vaccine shortly before being recruited (less than 24 h) by a general practice that was participating in the study, and participants or their parents/legally acceptable representative who the investigator believed could and would comply with the requirements of the study protocol. Persons already vaccinated with any other H1N1 pandemic vaccine before study enrolment and any child in care were excluded from participation. Written informed consent was provided by the participant or participant's parent or legally acceptable representative. A subset of the participants, who had at least one non-missing data for at least one solicited symptom, was asked to be a part of the reactogenicity cohort. Diary cards for assessment of reactogenicity were provided to participants in the reactogenicity cohort.
Participants were classified according to their risk of complications from influenza infection according to the definitions of the UK Department of Health:13 immunocompromised, at-risk, or healthy participants. Immunocompromised participants were those who reported immunosuppression at the administration of the first dose of vaccine. At-risk participants were participants who were not classified as immunocompromised and reported any of the following conditions at the administration of the first dose: spleen dysfunction or asplenia (defective or absent splenic function, respectively); chronic respiratory disease, including asthma; chronic neurological diseases and neurodevelopmental disorders; chronic renal disease; chronic liver disease; metabolic disease; immune system disorders; chronic haematological disorders or gastrointestinal disorders. Pre-existing conditions were reported by the participants at the time of enrolment based on medical history. All other participants were classified as healthy participants.
Criteria for evaluation
The primary endpoint was MAEs occurring within 31 days (D0–D30) after any dose. The secondary endpoints were solicited local (pain, redness and swelling) and general (children <5 years: fever, irritability, drowsiness, loss of appetite; participants ≥5 years: fever, headache, fatigue, gastrointestinal symptoms, joint pain, muscle ache, shivering and sweating) AEs self-reported during a 7-day follow-up period (D0–D6) after any dose, and SAEs and AESIs occurring within 181 days (D0–D180) after any dose. As recommended by the CHMP, the safety database was searched for all AESIs corresponding to the recommended preferred terms (PTs) or narrow Standardised Medical Dictionary for Regulatory Activities (MedDRA) queries (SMQs).10 Potential cases were identified according to available case definitions such as those developed by the Brighton Collaboration (http://www.brightoncollaboration.org) or medical judgment. A medically qualified person evaluated all cases reported for diagnosis ascertainment to identify confirmed cases of interest among all the potential cases identified. The medical evaluation of diagnosis certainty had three possible outcomes for each potential case:
-
Diagnosis confirmed (confirmed AESI),
-
Reported without sufficient information to conclude on diagnosis certainty, or
-
Diagnosis excluded (non-AESI).
Cases with a confirmed diagnosis and cases reported without sufficient information to conclude on diagnosis certainty were included in the observed-to-expected (O/E) analyses of AESIs, with the exception of two cases of anaphylactic shock that were related to concomitant medications.
The investigators assessed some of the reported AEs as possibly related to the vaccination, and general descriptive information on these related AEs is provided here. However, to increase sensitivity, all analyses included all reported AEs, irrespective of whether or not they were considered vaccination-related, as per the investigator's assessment.
Statistical analysis
The sample size was determined based on the recommendations of the EMA for postauthorisation evaluation of medicines for human use.10 The target population consisted of at least 9000 participants vaccinated according to the national vaccination programme at participating general practices. According to the EMA power estimations, ‘a total sample size of 9000 participants would be able to rule out at 95% confidence events (MAEs, SAEs and AESIs) occurring with a frequency of 1 per 3000 if no event is observed (provided that the event occur in all age categories)’.
Demographic characteristics were summarised by descriptive statistics. The incidence of solicited AEs in the reactogenicity subset, and the proportion of unsolicited AEs, SAEs, MAEs and AESIs in the total vaccinated cohort were calculated along with the associated 95% confidence intervals (CIs) using an exact method. MAEs, SAEs and AESI were categorised according to the MedDRA PT. Missing data were not replaced for the analysis of solicited symptoms. Analysis of MAEs, SAEs and AESIs included all vaccinated participants, and participants that did not report the event were considered as participants without the event. Incidences were computed for the overall population, per age group, risk groups and for pregnancy status. The following age groups were considered for the analysis: <2, 2−4, 5−9, 10−17, 18−44, 45−60 and >60 years. Observed-to-expected analyses were performed for AESIs and fatalities. In order to take the age distribution of the study population into account, an age-stratified expected number of cases was calculated. The observed incidences for AESIs within 31 and 181 days following the first dose were compared to expected incidences available for convulsion,15 optic neuritis,16 Bell's palsy,17 GBS18 and multiple sclerosis for demyelination.19 The expected rate was age-stratified, and the standardised incidence ratio (SIR) was calculated as observed/expected. SIR was presented by age group and overall, with 95% CIs based on the CI of the numerator. As only one case of GBS was identified in a single male participant, the observed number of cases was compared to the expected number of cases for males only. Expected mortality rates were retrieved from the Office for National Statistics, UK.20 The standardised mortality ratio (SMR) was calculated for the follow-up periods of 31 and 181 days after each dose as observed incidence rate (IR) divided by expected IR. SMR was presented by age group and overall, with 95% CIs based on the CI of the numerator. The date of the event was defined as the date of death and not the date of onset of the associated AE. For any participants who were lost to follow-up, a request was sent to the National Health Service (NHS) Information Centre Medical Research Department in order to identify any fatality that was not recorded.
The software used for all statistical analyses was SAS (Statistical Analysis System) V.9.2.
Results
Demographics
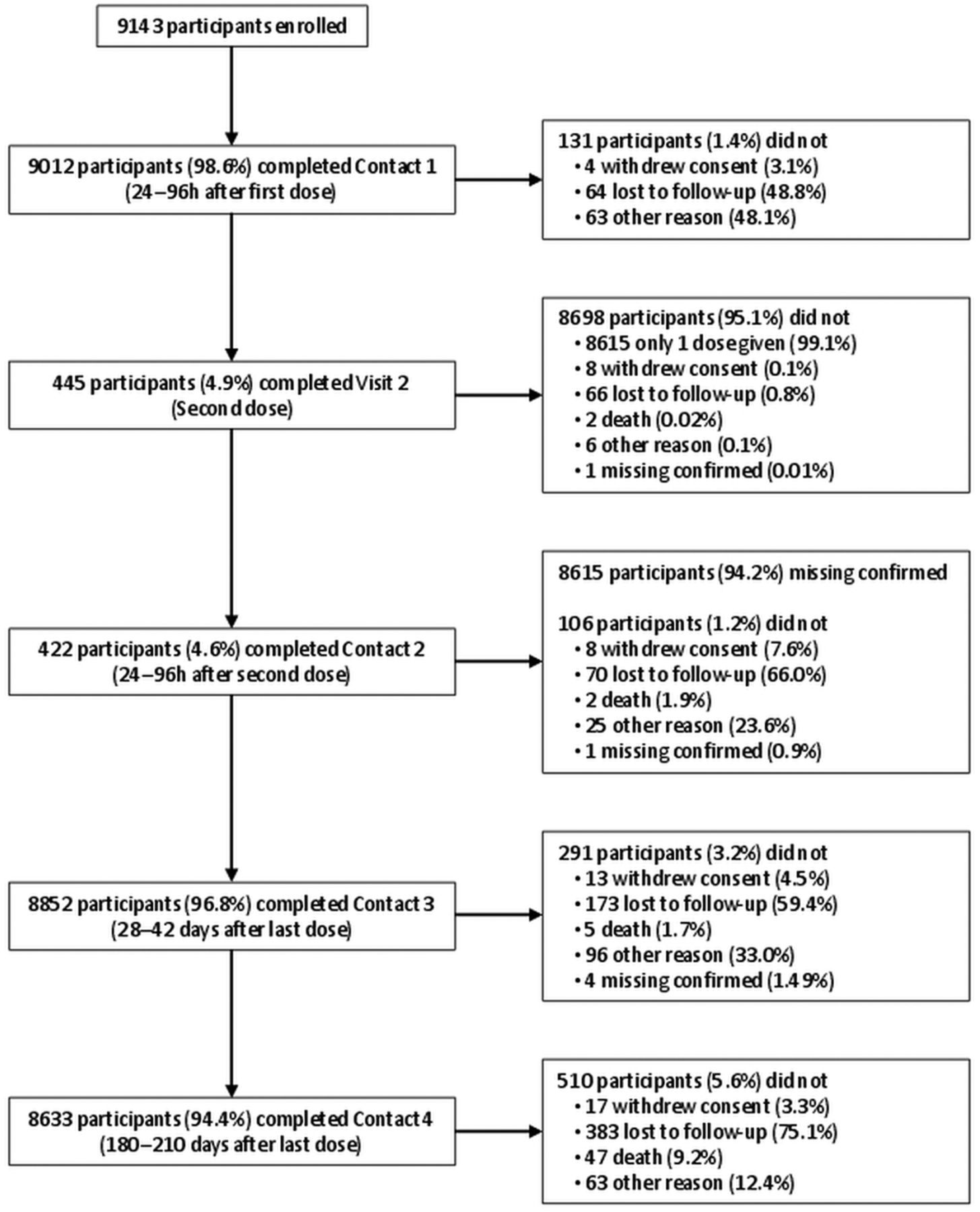
From the MRC GPRF, 120 English general practices were asked to partake in the study. Of these, 87 general practices participated and these were largely distributed throughout England. A total of 9215 participants were enrolled, and data for analysis were available on 9143 participants (Study cohort). Further, 72 participants were eliminated for not complying fully with the written informed consent process. The mean (±SD) age of the study cohort was 54.7±20.2 years (range 7 months to 97 years) and 51.1% were female (table 1). The majority (80.8%) of participants were in the non-immunocompromised and at-risk group, 6.3% were immunocompromised and 12.8% were healthy participants. In all, 94.4% (N=8633/9143) of the participants completed the 6-month follow-up. Reasons for non-completion of the study are detailed in figure 1.
Demographic characteristics of the study cohort and the reactogenicity cohort

Flow diagram depicting the completion of study contact points with the reasons for discontinuation. Participants with contacts not performed for other reasons could have had the following contacts. If only one dose of vaccine was given, Contact 2 was considered ‘Missing confirmed’.
Reactogenicity
The reactogenicity analysis included 682 participants (52.8% females) (table 1). Overall, the most frequently reported solicited local AE was injection site pain (children ≤17 years: 79.5%, adults: 78.3%) followed by injection site redness (children: 49.6%, adults: 19.8%) for both age groups (figure 2A). The median duration of local symptoms ranged between 2 and 5 days for any symptom. In children, the incidence of local symptoms was higher in at-risk participants than healthy children, especially for swelling (43.4% (32.1 to 55.3) vs 19.5% (8.8 to 34.9)) (table 2). In adults, local pain was more frequently reported by healthy participants (80.0%) and participants at risk (78.5%) than immunocompromised participants (73.0%). Local redness (27.0%) and swelling (21.6%) were more frequently reported in immunocompromised participants than in healthy participants or participants at risk (table 2). The median duration of local symptoms was somewhat longer in immunocompromised participants (4.0–4.5 days) compared to healthy participants (2.0–3.0 days) and participants at risk (3.0 days).
Proportion (%) of participants with solicited local and general adverse events (AEs) reported within the 7-day postvaccination period (Reactogenicity cohort N=682)

{kind=link}
{kind=link}
Solicited local (A) and general (B) adverse events reported during a 7-day follow-up period after any dose (Reactogenicity cohort N=682). The general symptoms of drowsiness, irritability and loss of appetite were only assessed in children <5 years, whereas fatigue, gastrointestinal, headache, joint pain, muscle aches, shivering and sweating were assessed in children aged 5–17 years and in adults. Fever was defined as an oral or axillary temperature of ≥37.5°C (99.5°F) or a rectal temperature of ≥38.0°C (100.4°F). Data are shown as the percentage of participants reporting the symptom with 95% CI.
In children <5 years of age, irritability was the most common solicited general AE (61.8%; figure 2B). Most of the solicited general AEs were reported more often for children aged <5 years who were considered healthy compared to those at risk (table 2). Myalgia (muscle aches) was the most common solicited general AE in children aged 5–17 years (61.9%) and adults aged >17 years (46.9%). The overall proportion of participants with Grade 3-solicited symptoms did not exceed 7.7%. In children aged 5–17 years, most of the symptoms were commonly observed in at-risk children, except for fever which was more frequently observed in healthy children (28.6% vs 14.3%) and for joint pain for which there was no difference between the groups (28.6% in both groups). In adults, the reactogenicity profile was generally highest in the immunocompromised participants compared to the healthy participants and participants at risk (table 2). In all age groups, the median duration of Grade 3-solicited general symptoms ranged between 1 day and 2 days.
MAEs, SAEs and AESIs
At least one MAE was reported by investigators for 13.3% (1219/9143) of participants within the 31-day postvaccination period (table 3). The most frequently reported MAEs were associated with ‘infections and infestations’. Lower and upper respiratory tract infections were the most frequently reported event PTs. A higher proportion of MAEs (any symptom) was reported in the immunocompromised participants (18.5%) compared to at-risk (13.0%) and healthy (13.3%) participants. A total of 154 participants experienced at least one MAE assessed by investigators as possibly related to vaccination, with the most frequently reported event PTs being lower respiratory tract infection (16/9143), upper respiratory tract infection (10/9143) and cough (10/9143).
Most frequently reported (≥9 cases) medically attended adverse events (MAEs) within the 31-day postvaccination period
At least one SAE was reported for 4.5% (411/9143) of participants in the study cohort during the 181-day postvaccination period with pneumonia (16 cases), lower respiratory tract infections (13 cases) and asthma (13 cases), the most frequently reported event PTs (table 4). Of these, 1.2% (113/9143) of participants reported at least one SAE during the 31-day postvaccination period, with lower respiratory tract infection (0.07%, 6/9143) being the most frequently reported event PT. Eleven participants experienced at least one SAE assessed by investigators as possibly related to vaccination, with asthma/asthmatic crisis being the most frequently reported event PT (3/9143).
Most frequently reported (≥5 cases) serious adverse events (SAEs) during the 181-day postvaccination period (N=9143)
During the 181-day postvaccination period, 22 participants reported 26 potential AESIs. After medical review, only 18 AESIs (including confirmed cases and cases for which there was insufficient information to confirm the certainty of diagnosis) in 14 participants were considered for the O/E analyses (table 5). These 14 participants included: 1 participant <2 years old, 1 from the 10−17 years age group; 1 from the 18−44 years age group; 3 from the 45−60 years age group and 8 from the >60 years age group. The most frequently reported AESI was convulsion: 11 episodes of convulsion occurring in 8 participants. For participants with more than one episode of convulsion, only the first occurrence after vaccination was included in the analyses. AESIs not included in the analyses were two cases of anaphylactic reaction experienced by two participants, which occurred at 69 and 145 days after vaccination and were causally associated to other medications (atracurium besylate in one case and terbinafine in the other case); one case of polymyalgia rheumatica which was not associated with vasculitis; and five cases of circulatory collapse in five elderly participants. These five cases were excluded as anaphylaxis, as they were assessed by the investigators as being associated to the patients’ coexisting cardiovascular diseases.
Adverse events of special interest (AESIs) reported within the 181-day postvaccination period (N=9143)
There were 53 deaths (0.58%) reported during the entire study period, with an additional three cases retrieved from the NHS Information Centre Medical Research Department. In particular, 41 deaths occurred during the 181-day period after vaccination; one additional case was retrieved from the NHS Information Centre Medical Research Department, corresponding to an incidence mortality rate of 940/100 000 person-years (95% CI 675 to 1275). None of the fatalities reported (40 cases) were considered by the investigator as related to vaccination, while the one additional fatality was assessed by a GlaxoSmithKline safety physician who considered that there was no reasonable possibility that the fatal event was related to vaccination, but rather that it was related to the participant's medical conditions. The majority of fatality reports described participants older than 60 years (50/56, 89.3%) and were identified as possibly associated with the presence of pre-existing chronic medical conditions. No fatalities were reported in participants younger than 45 years of age.
O/E analyses
The observed number of fatalities was below the expected number of fatalities (SMR 0.45; (95% CI 0.32 to 0.61)). There were no reports suggestive of non-infectious encephalitis and vaccination failure and no confirmed reports of vasculitis or vaccine-related anaphylactic reaction. According to the O/E analysis, the incidence of AESI was higher than expected for two AESIs. The first AESI was neuritis, for which a single case occurred within 30 days (SIR 65.51 (1.66 to 365.01)). This event was not considered serious. It was reported in one non-immunocompromised at-risk 86-year-old man with no relevant past medical history. On the day of vaccination, the participant experienced cervical stiffness and paresthesia in the left hand and was diagnosed with neuritis (not specified otherwise). No clinical details or relevant diagnostic test results were provided by the investigator. The second AESI was convulsions with two cases reported within the 30 days (3.84 (0.47–13.89)), but it was only significant for the 181-day interval (2.65 (1.14 to 5.22)).
Discussion
Statement of principal findings
This prospective observational study was set up in time to enrol the first participant when the mass vaccination campaign began in the UK. Overall, the target recruitment was exceeded for both the study cohort and the reactogenicity cohort. Only a limited number of participants were lost to follow-up (<6%). The solicited AEs reported were primarily common local and general symptoms: injection site-related AEs, irritability in young children and muscle aches in older children and adults. MAEs were reported for 1219 participants during the 31-day postvaccination period. The most frequently reported MAEs and SAEs were consistent with events anticipated to be reported by the populations under study, particularly during the winter season, that is, respiratory tract infections. The observed number of fatalities was below the expected number of fatalities. There were no reports suggestive of non-infectious encephalitis and vaccination failure, and no confirmed reports of vasculitis or vaccine-related anaphylactic reaction were received.
Confirmed cases of AESIs were rare (0.15%). The observed number of cases of Bell's palsy, GBS and demyelination was below the expected number. The observed number of convulsions was higher than expected for the 181-day interval, but not for the 31-day interval; the lack of temporal association with vaccination is reassuring. The observed number of neuritis cases was higher than expected for the 30-day interval, considering that only one case was retrieved. This event occurred in a non-immunocompromised and at-risk 86-year-old man with no relevant past medical history. On the day of vaccination, the patient experienced neck stiffness and paraesthesias of his left hand. No clinical details or relevant diagnostic test results were provided, and the final diagnosis was neuritis. In general, the O/E analysis was overly sensitive, as both prevalent cases and cases reported without sufficient information to conclude on diagnosis certainty were included. Furthermore, no correction was made for the multiplicity of comparisons.
Strengths and weaknesses of the study
General practices are the primary contact point for persons in the UK to access the National Health Service. The general practices were able to provide an almost complete overview of all medical events that occurred throughout the study,14 so an almost complete ascertainment of the safety profile of the AS03 adjuvanted H1N1 (2009) pandemic influenza vaccine is the main strength of this study. The second strength of this study was the number of participants (ie, over 9000) enrolled, which exceeded the sample size recommended by EMA for pharmacovigilance activities concerning pandemic vaccines.10 Nevertheless, there are some limitations in this study. First, no sample size estimations of the number of participants that should have been enrolled in each risk group (immunocompromised, at-risk and healthy participants) were performed. Thus, it is difficult to ascertain whether the analysis reported here was sufficiently powered to adequately assess safety outcomes such as reactogenicity and MAEs in the general UK population. Additionally, the majority of participants involved in the study (81%) were classified as at-risk, according to the definitions of the UK Department of Health,13 and consequently enrolled in the at-risk group, resulting in a sample structure that differed from that of the general population. Second, a related limitation of this study is that the sample size may not be large enough for the assessment of the potential for the vaccine to be associated with rare AEs such as autoimmune diseases. Another limitation is that there was no comparator group, so proportions of observed outcomes were compared with the available background rates from the general population derived from the literature.
Strengths and weaknesses of the study in relation to other studies
The reactogenicity and safety profiles of healthy participants were generally comparable to those observed in other trials on the H1N1 pandemic7 ,21–25 and H5N1 prepandemic8 vaccines. However, in the <5 years group, all general symptoms tended to be higher when compared to an H1N1 pandemic vaccine clinical trial (eg, irritability 46.2% vs 61.8% in this study).26 Also, in the <5 years group, drowsiness and irritability tended to be higher when compared to an H1N1 pandemic vaccine25 and an H5N1 prepandemic vaccine clinical trial (for instance, drowsiness 24.5% vs 38.2% and irritability 36.7% vs 61.8%).27
There were 18 AESIs reported with the most common AESI being 11 episodes of convulsions in eight participants. Five of these participants had a medical history of convulsion or epilepsy and, according to the study's investigators, the convulsive episode was triggered by other possible causes (eg, trauma, acute infection, alcohol consumption or lack of compliance with treatment). Febrile convulsion was only reported in one participant, a healthy 8-month-old girl child. The remaining participants experienced a first convulsive episode occurring 38 and 123 days, respectively, after vaccination, with no apparent cause. The incidence of convulsions, in particular febrile convulsions, has recently received much attention after an increased incidence of severe febrile convulsions in young children led to the suspension of the 2010 seasonal influenza vaccination programme in Western Australia.28 Further investigation into the cause of these convulsions showed that it was due to vaccination with one particular brand of trivalent seasonal influenza vaccine and not associated with prior vaccination with the seasonal influenza or 2009 H1N1 pandemic vaccine.29 Indeed, a recent study did not demonstrate an association between an increased risk of convulsions and vaccination with seasonal trivalent influenza vaccines (over a 10-year surveillance period) or the AS03-adjuvanted pandemic H1N1 vaccine in 2009–2010.30
Another AESI of particular interest is demyelination. Some forms of demyelination attack the central nervous system (the main example being multiple sclerosis), while others affect the peripheral nervous system (the main example being GBS, which was analysed separately as AESI). There was one case of GBS reported in this study, diagnosed as a possible mild GBS, occurring 106 days after vaccination in a 78-year-old non-immunocompromised and at-risk man who had a pre-existing medical condition of polyneuropathy (not otherwise specified). A previous mass vaccination campaign that ended in 1976 against swine influenza in the USA was suspended owing to the significantly increased rate of GBS in adults of all ages.31 Although no increased risk of GBS following influenza vaccination was detected during the two subsequent seasonal influenza seasons,32 ,33 the incidences of GBS and similar AEs following mass vaccination campaigns are still a concern. Although a systemic review of the meta-analysis of clinical trials assessing the effectiveness of the pandemic influenza A/H1N1 2009 vaccine did not detect any cases of GBS following vaccination,34 a preliminary analysis by the Centers for Disease Control in the USA suggested a significant association between the 2009 H1N1 vaccination and GBS.35 Recent studies performed in several European countries reported no increased risk of GBS with the pandemic influenza A/H1N1 2009 vaccine.36 ,37 It has been a matter of debate whether vaccination may have the potential to exacerbate pre-existing relaxing-remitting conditions such as multiple sclerosis. This study was not adequately powered to rule out a clinically relevant association between the 2009 H1N1 vaccination and a pre-existing relaxing-remitting condition. In our study, there was one participant who had a pre-existing secondary progressive multiple sclerosis that experienced a possible aggravation or flare-up occurring 62 days after vaccination. Multiple sclerosis relapse has been considered when assessing the evidence of a possible association with influenza vaccines. Clinical studies with cohorts of multiple sclerosis patients generally concluded that influenza vaccination did not appear to be associated with an increased risk of multiple sclerosis relapse.38–41
Conclusions
This study has shown that the 2009 pandemic influenza vaccine adjuvanted with the AS03 Adjuvant System showed clinically acceptable reactogenicity and safety profiles in all age and risk groups studied. There were limited safety data available regarding the safety of this vaccine in both children and adults before the outbreak of the pandemic. Thus, the experience acquired with this vaccine will be of benefit for the development of future vaccines against pandemic influenza outbreaks.
Acknowledgments
This study was successfully managed by Maureen Hitschfield (freelance, on behalf of GlaxoSmithKline Vaccines) and was made possible by the dedication of all persons involved in the study conduct. We are grateful to Claire Marie Seymour and Adriana Rusu (XPE Pharma & Science), who provided writing assistance on behalf of GlaxoSmithKline Vaccines, and to Géraldine Drevon (GlaxoSmithKline Vaccines) who provided editorial assistance and manuscript coordination. We thank all the study participants and the general practitioners, practice nurses, and practice staff in the MRC General Practice Research Framework and the National Institute of Health Research Primary Care Research Network who took part in this study, including Alistair Adey, Shana Agarwall, Ayotunde Ajala, Martin Armer, Sally Barnard, Mike Beer, Jonathan Brunskill, Lynne Burke, Tom Cahill, Jonathan Chapman, Chuba Chigbo, Margaret Chung, Warwick Coulson, Allison Crumbie, Rhodri Davies, Andrew Down, David Edwards, Mark Field, Ian Fitzsimmons, Peter Franklin, Ahmet Fuat, Matthew Gaw, Bozena Gorecka, Elizabeth Green, Peter Greenaway, Michael Gumbley, Nicola Harding, John Harrison, Geraldine Hogan, Kate Hopkins, Nigel Hume, Ewart Jackson-Voyzey, Ian James, John James, Rupert Jones, Allison Julie, Debra Kaye, Vijay Koshal, Rajan Kuriakose, Denise Linscott, Pippa Lally, David Lawson, Mark Lee, Briony Lodge, Virendra Mittal, Elizabeth Mackenzie, Vinay Mehra, Michael Moore, Joanna Nash, Macgy Neil, Ian Morrey, Carolyn Paul, Basil Penney, Jonathan Price, Sue Pritchard, Doug Redpath, Simon Royal, Peter Saul, David Seamark, Steven Shapiro, Justin Smith, Amrit Takhar, Julian Thompson, Mark Thompson, Phil Townsend, Freya Yoward, Helen Yoward, Simon Wetherell, Eric Wilkinson, Varendar Winayak. We are grateful to Daniel De Palmenaer and Cathlijn Leenaars (GlaxoSmithKline Vaccines) for preparation of study-related documentation. We would like to address special thanks to Felix Arellano and David Vaughn (GlaxoSmithKline Vaccines) for a critical reading of the manuscript and very helpful suggestions.
References
Footnotes
-
Contributors VB, IN and DR conceived and designed the study. IN acquired the data. FH carried out statistical analysis. DR participated in the statistical analysis. All authors conceived the paper and participated in the analysis and interpretation of the data. They were involved in the drafting of the article or revising it critically for important intellectual content, as well as in the final approval of the manuscript. They also had full access to the data and had final responsibility to submit for publication. VB is the guarantor.
-
Funding This work was supported by GlaxoSmithKline Biologicals SA who took responsibility for all costs associated with the development and publishing of the present manuscript.
-
Competing interests IN has received support from GlaxoSmithKline Biologicals SA through a grant as part of the MHRA postsafety surveillance study on MRC GPRF; FT, DR, FH and VB are employees of the GlaxoSmithKline group of companies; FT and VB report ownership of GlaxoSmithKline stock options.
-
Patient consent Consent was not obtained from the participants but the presented data are depersonalised and the risk of identification is low.
-
Ethical approval All participants provided written informed consent before taking part. The study protocol and informed consent forms were reviewed and approved by a national Independent Ethics Committee.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement There are no additional data available.
-
Author footnote Pandemrix is a trademark of GlaxoSmithKline group of companies.
Linked Articles
- Correction