Article Text

Abstract
Objectives Single genetic loci offer little predictive power for the identification of depression. This study examined whether an analysis of gene–gene (G × G) interactions of 78 single nucleotide polymorphisms (SNPs) in genes associated with depression and age-related diseases would identify significant interactions with increased predictive power for depression.
Design A retrospective cohort study.
Setting A survey of participants in the Wisconsin Longitudinal Study.
Participants A total of 4811 persons (2464 women and 2347 men) who provided saliva for genotyping; the group comes from a randomly selected sample of Wisconsin high school graduates from the class of 1957 as well as a randomly selected sibling, almost all of whom are non-Hispanic white.
Primary outcome measure Depression as determine by the Composite International Diagnostic Interview–Short-Form.
Results Using a classification tree approach (recursive partitioning (RP)), the authors identified a number of candidate G × G interactions associated with depression. The primary SNP splits revealed by RP (ANKK1 rs1800497 (also known as DRD2 Taq1A) in men and DRD2 rs224592 in women) were found to be significant as single factors by logistic regression (LR) after controlling for multiple testing (p=0.001 for both). Without considering interaction effects, only one of the five subsequent RP splits reached nominal significance in LR (FTO rs1421085 in women, p=0.008). However, after controlling for G × G interactions by running LR on RP-specific subsets, every split became significant and grew larger in magnitude (OR (before) → (after): men: GNRH1 novel SNP: (1.43 → 1.57); women: APOC3 rs2854116: (1.28 → 1.55), ACVR2B rs3749386: (1.11 → 2.17), FTO rs1421085: (1.32 → 1.65), IL6 rs1800795: (1.12 → 1.85)).
Conclusions The results suggest that examining G × G interactions improves the identification of genetic associations predictive of depression. 4 of the SNPs identified in these interactions were located in two pathways well known to impact depression: neurotransmitter (ANKK1 and DRD2) and neuroendocrine (GNRH1 and ACVR2B) signalling. This study demonstrates the utility of RP analysis as an efficient and powerful exploratory analysis technique for uncovering genetic and molecular pathway interactions associated with disease aetiology.
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
Article summary
Article focus
Single genetic loci offer little predictive power for the identification of depression. This study examined whether an analysis of G × G interactions of SNPs in genes associated with depression and age-related diseases would identify significant interactions with increased predictive power for depression.
Key messages
Using a classification tree approach (RP), we identified a number of candidate G × G interactions associated with depression. After controlling for G × G interactions by running LR on RP-specific subsets, every split became significant and grew larger in magnitude. Four of the SNPs identified in these interactions were located in two pathways well known to impact depression: neurotransmitter (ANKK1 and DRD2) and neuroendocrine (GNRH1 and ACVR2B) signalling.
Strengths and limitations of this study
Our results suggest that examining G × G interactions improves the identification of genetic associations predictive of depression. This study demonstrates the utility of RP analysis as an efficient and powerful exploratory analysis technique for uncovering genetic and molecular pathway interactions associated with disease aetiology.
Introduction
Depression is a widespread mental disorder associated with a host of undesirable health, social and economic outcomes. One in six Americans is diagnosed with depression in his or her lifetime.1 While many environmental factors—such as socioeconomic status, childhood abuse and major life events—have important ties with depression, so too does gender and many genetic and epigenetic factors, making the disorder heterogeneous in nature.2 Another major risk factor for depression is age, with depression reaching its highest levels in adults aged 80 years and older.3
It has been demonstrated from twin studies that genetic factors typically account for 40%–70% of the risk for developing major depressive disorder, and adoption studies have confirmed the role of genetic risk factors in the development of major depressive disorder (see Zubenko et al4 and references therein). Genetic studies, including recent genome-wide association studies (GWAS), have identified genetic alterations in over 50 genes known to be associated with depression.5 However, individually, the genetic alterations found within these genes (primarily single nucleotide polymorphisms (SNPs)) have little predictive value. There is a similar lack of predictive value from GWAS of other major age-related diseases.6
Given this lack of predictive power among individual genetic alterations for depression together with the complex nature of ageing-related diseases, it would seem prudent to examine epistatic effects on this age-related condition. In this respect, we have previously demonstrated that gene-gene (G × G) interactions greatly modulate risk for complex age-related diseases.7 ,8 Recent studies of depression also have identified epistatic effects. In particular, associations have been identified between BDNF Val66Met (brain-derived neurotrophic factor; rs6265) and 5-HTTLPR (serotonin transporter-linked promoter region9); GSK3B rs6782799 (glycogen synthase kinase 3β), BDNF rs7124442 and BDNF Val66Met10; BDNF Val66Met and SNPs in NTRK2 (neurotrophic tyrosine kinase receptor 211); and 5-HTTLPR short allele and a chromosome 4 gene.12 The machine learning tool recursive partitioning (RP) has recently been used by Wong et al13 to assess complex G × G interactions in depression. Wong et al note that RP is useful in that it quickly explores high-dimensional data for non-linear effects that are non-biased and easily interpretable.
The goals of this study were therefore to (1) explore G × G interactions that might better predict the genetic factors involved in the aetiology of depression and (2) to further demonstrate the utility of machine learning algorithms (RP) to identify genetic interactions. Using genotypic data from the Wisconsin Longitudinal Study (WLS), we identified associations between dopaminergic genes and depression in men and women, as well as G × G interactions involving neuroendocrine signalling pathways, with increased significance compared with single genetic associations.
Methods
Study participants and surveys
Data were collected from the WLS, a random sample originally comprised 10,317 men and women who graduated from Wisconsin high schools in 1957. Later in 1977, the WLS began interviewing one randomly selected sibling of each graduate, when possible. The cohort reflects the ancestral makeup of the late-1950s Wisconsin population in that participants are almost entirely non-Hispanic white men and women. In general, the sample is broadly representative of older white Americans with at least a high school education.14 Further characteristics of the WLS cohort may be found in detail elsewhere.15 Health and psychological well-being phenotypic data were taken from mail and phone surveys given in 2004–2005. Inclusion criteria for depression included any member of the WLS cohort who was depressed according to the Composite International Diagnostic Interview–Short-Form. Individuals who answered YES to the question ‘Have you ever had a time in life lasting two weeks or more when nearly every day you felt sad, blue, depressed, or when you lost interest in most things like work, hobbies, or things you usually liked to do for fun?’ and whose depression was not caused by alcohol, drugs, medications or physical illness were asked further depression symptom questions. Symptom questions asked whether the 2-week period was accompanied with (1) any weight loss, (2) trouble sleeping, (3) feeling tired, (4) feeling bad upon waking, (5) losing interest, (6) trouble concentrating or (7) thoughts about death. Those answering YES to three or more of these symptom questions were classified as having depression.16 Those answering YES to two or fewer symptom questions and all those answering NO to the initial stem question were classified as controls.
Genotyping
Seven thousand one hundred and one participants (4569 graduates and 2532 siblings) provided saliva samples in Oragene DNA sample collection kits (DNA Genotek, Kanata, Canada) from which DNA was extracted and genotyped for 78 SNPs that were selected based on their association with depression and age-related conditions and diseases (see supplementary information 1). Genotyping was performed by KBioscience (Hoddesdon, UK) with use of a homogeneous Fluorescent Resonance Energy Transfer technology coupled to competitive allele-specific PCR. All SNP genotypes described in our results were in Hardy–Weinberg equilibrium and their frequencies matched those reported in the literature for European samples.
Statistical analysis
Analyses were limited to the 4811 pooled graduates and siblings for whom we had depression and genotype information (note: individuals with more than 10% missing genotype data were not included). The average age among this sample was just younger than 65 years in 2004. Eighty per cent were married, and the average amount of post-high school educational attainment was 2 years. Median household income in 1993 was $56 700.
Recursive partitioning
RP is a data mining tool for revealing trends that relate a dependent variable (depressed vs non-depressed) to various predictor variables (SNPs). Zhang and Bonney17 have shown how RP can be used in genetic association studies to identify disease genes. RP helps control for heterogeneity in the population and confounding factors by allowing for the segregation of the sample population according to any condition. Thus, RP is a useful way to handle complex data sets that might confound regression analysis due to the complexity of the relationship between the independent and dependent variables and due to missing information.
RP classification trees (using R package rpart) were used to identify potential interactions among the 78 SNPs in relation to depression. The trees split the data along branches according to the criteria determined by the rpart package algorithm, which is originally based off the work of Breiman's classification and regression trees algorithm.18 Basically, the classification and regression trees algorithm first considers all depressed and non-depressed subjects pooled together in a heterogeneous root node. Based on considering every possible ‘yes–no’ binary partition that can be made by each independent variable, the single split, which maximises homogeneity between the two resulting subnodes as compared with the root node, is made. Each subnode can then be treated independently as a new root node for all subsequent splits, and the pattern continues until every subject constitutes a terminal node, resulting in a very large and complex tree. A 10-part cross-validation procedure seeking to minimise misclassification and complexity determines optimal pruning. See Therneau and Atkinson19 for specific details of the rpart package. Priors were set to 0.5, 0.5. The use surrogate parameter was set to 0 so that subjects missing the primary split variable do not progress further down the tree, and maxsurrogate was set to 0 to cut computation time in half. The threshold complexity parameter was set to 0.01. Tree nodes were re-created in Microsoft Visio to display percentage depressed and the default number of controls/cases as presented by the rpart.
Logistic regression
Variables found in association with depression based on RP analysis were considered in single-factor logistic regression (LR) models, separate by gender, using the specific dichotomous splitting of genotypes as designated by RP trees. Regression models for all seven SNP splits were first run on the full data set to represent single main factor effects. Then, each split was run on the respective subset of data as represented by the preceding RP split criteria. Thus, we attempt to mirror RP splits within a more formal LR framework in order to measure the significance of interactions presented by the trees. Multiple testing of 78 SNPs in RP for both men and women followed by 14 LR models resulted in a modified false discovery rate (FDR) significance level of 0.009.
Results
Of the 4811 participants (2464 women and 2347 men) under examination in this study, we identified 713 participants (481 women and 232 men) with depression (14.8%). Given that the independent variable gender (when included as a factor in the full data set) was the primary split on RP trees; that women are over two times as likely to be diagnosed with depression than men and since the female aetiology of depression has been reported to be associated with unique social, psychological, and biological factors,20 all subsequent analyses were performed by gender.
Recursive partitioning analysis
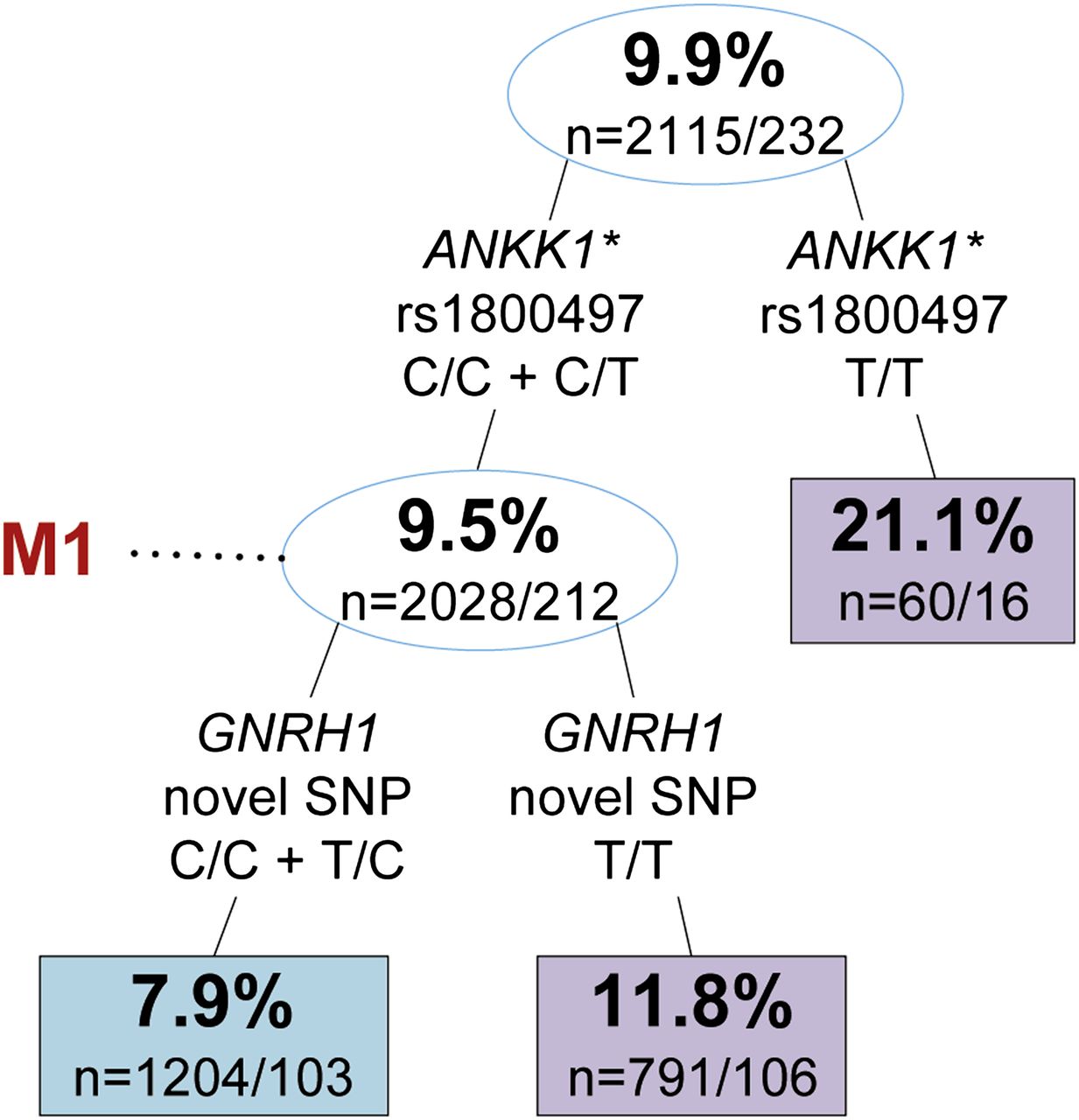
To examine multigene interactions for association with depression, we screened our data set using RP. The two-factor RP tree (ANKK1/GNRH1) was the optimised pruning for men (figure 1), while the five-factor tree (DRD2/APOC3/ACVR2B/FTO/IL6) was the optimised pruning for women (figure 2). For more detailed information on the seven SNPs found by RP, see supplementary information 2.

Recursive partitioning tree of Composite International Diagnostic Interview–Short-Form depression in men of the Wisconsin Longitudinal Study. Upper and lower numbers in nodes represent the percentage of participants with depression and the number of controls/cases in that node, respectively. Blue and purple boxes/circles indicate lower and higher rates of depression relative to the primary node, respectively. Split information indicates gene, single nucleotide polymorphism (SNP), and genotype criteria, respectively. M1 is subset of data referenced in table 1. Sensitivity: 0.526, specificity: 0.598, accuracy: 0.591. Due to missing genotype information, we lose approximately 1.5% of participants per split. *rs1800497 is historically known as the DRD2 Taq1A allele.

{kind=link}
{kind=link}
Recursive partitioning tree of Composite International Diagnostic Interview–Short-Form depression in women of the Wisconsin Longitudinal Study. Upper and lower numbers in nodes represent the percentage of participants with depression and the number of controls/cases in that node, respectively. Blue and purple boxes/circles indicate lower and higher rates of depression relative to the primary node, respectively. Split information indicates gene, single nucleotide polymorphism (SNP), and genotype criteria, respectively. F1–F4 are subsets referenced in table 1. Sensitivity: 0.607, specificity: 0.563, accuracy: 0.572. Due to missing genotype information, we lose approximately 1.4% of participants per split.
The best overall split for men was ANKK1 rs1800497 (historically known as the DRD2 Taq1A allele), where the incidence of depression increased 2.2-fold in those with no C-alleles compared with those with one or two C-alleles. Considering interaction between ANKK1 and GNRH1 widened the disparity in incidence, where those with at least one C-allele in both ANKK1 rs1800497 and the novel SNP in GNRH1 had a 2.7-fold lower incidence than those without a C-allele in ANKK1 rs1800497.
For women, the best overall split was DRD2 rs2242592, where those with one or two C-alleles had 1.3-fold higher incidence of depression compared with those without any C-alleles. G × G interactions associated with the highest incidence of depression included DRD2 rs2242592 T/T + APOC3 rs45537037 T/T + ACVR2B rs3749386 C/C or T/T, accounting for a 1.4-fold increase in depression compared with baseline incidence.
Single main factor effects
Specific SNP interactions identified by RP were next analysed by LR (see table 1 for full data). The primary SNP splits in men and women were significant at the modified FDR level. Men with no C-alleles for ANKK1 rs1800497 had 2.55 times higher odds (p=0.001 (95% CI 1.44 to 4.51)) of depression compared with men with at least one C-allele. Women with at least one C-allele for DRD2 rs2242592 had 1.32 times higher odds (p=0.006 (95% CI 1.08 to 1.62)) of depression compared with women with no C-alleles. One other split reached nominal significance; women homozygous (C/C or T/T) for FTO rs1421085 had 1.32 times higher odds (p=0.008 (95% CI 1.08 to 1.62)) for depression than women with a heterozygous genotype. SNP splits of GNRH1, APOC3, ACVR2B, and IL6 did not significantly associate with depression.
Gene–gene interactions enhance predictability for depression
Specific SNP interactions identified by RP were next analysed by LR as RP-specific subsets (see table 1, RP-subsetted data). All five of the secondary and tertiary RP splits were found to be significant at the modified FDR level when considered as subsets. Among only men with at least one C-allele in ANKK1 rs1800497, those with no C-allele in the novel SNP of GNRH1 had 1.57 times higher odds (p=0.002 (95% CI 1.18 to 2.08)) for depression than men with one or two C-alleles. For the subset of women in the first right-hand split of figure 2, those homozygous for FTO rs1421085 had 1.65 times higher odds (p=0.0005 (95% CI 1.24 to 2.18)) for depression than women with a heterozygous genotype. For the remaining subset of women in the second right-hand split of figure 2, those homozygous for IL6 rs1800795 had 1.85 times higher odds (p=0.006 (95% CI 1.19 to 2.89)) for depression than women with a heterozygous genotype. For the subset of women in the first left-hand split of figure 2, those with no C-alleles for APOC3 rs45537037 had 1.55 times higher odds (p=0.004 (95% CI 1.15 to 2.09)) for depression than women with one or two C-alleles. For the subset of women in the second left-hand split of figure 2, those homozygous for ACVR2B rs3749386 had 2.17 times higher odds (p=0.001 (95% CI 1.37 to 3.44)) for depression than women with a heterozygous genotype.
Discussion
Using RP as a screening tool to find potential multigene interactions, followed by verification by LR, our data demonstrate that multigene interactions predict depression with a greater certainty than single main factor associations. RP provided us with primary dichotomous genotype splits in men and women (ANKK1 rs1800497 and DRD2 rs2242592, respectively) that were both significant in LR models at the modified FDR level (table 1). When considering the five subsequent RP splits over the entire data set with LR, only one reached a nominal level of significance (barely), which was FTO rs1421085 in women. However, after running LR on specific subsets of data according to the pattern of RP branches, every split was found to be significant and every OR grew larger (table 1; OR (before)→(after): male left: 1.43 → 1.57, female left 1: 1.28 → 1.55, female left 2: 1.11 → 2.17, female right 1: 1.32 → 1.65, female right 2: 1.12 → 1.85). Thus, RP provides two unique and important criteria: dichotomous genotype splitting instructions and G × G interaction patterns. These criteria go beyond the traditional single-factor SNP approach to genetic association studies and allow identification of important multigene pathways that more suitably characterise the aetiology of complex diseases.
The utility of RP and LR for identification of gene–gene interactions
With recent advances in genotyping allowing for high-dimensional SNP identification, it is now possible to examine genetic data sets for single main factor effects and also for G × G interactions. The requirement for G × G analyses as a better predictor of age-related diseases is obvious from the standpoint that humans are complex biological systems composed of numerous molecular interactions and from recent studies indicating disease risk is modulated by G × G interactions.7 Notwithstanding this, the development of analytical tools for the identification of G × G interactions has not kept pace with the technological advances in identifying genetic alterations among individuals. In this respect, we have previously used multifactor dimensionality reduction (MDR), LR and linkage disequilibrium (LD) to identify G × G interactions among a small set of SNPs.7 However, large data sets require a screening tool to identify potential multigene interactions. In this study, we have used RP to screen for multigene interactions, a data mining technique that is currently underused in genetic studies. RP serves as an efficient and powerful exploratory analysis technique, especially when looking for interactions in data sets with a large number of independent variables. This screening allows for the identification of G × G interactions (with greater explanatory power) that might otherwise not have been identified and that can then be confirmed using more traditional statistical techniques. As illustrated in this paper, this data mining methodology has the advantage of identification of genetic interactions between pathways involved in the aetiology of depression, in keeping with the etiological heterogeneity of this disorder (see later).
Our study provides proof of principle for the use of RP in higher dimensional analyses such as GWAS, where a comprehensive list of SNPs may fully explore genetic predisposition to depression and other age-related disease. The WLS is an ideal candidate for future GWAS studies, given its large sample size, rich covariate composition and longitudinal nature.
In this genetic study, we aimed to identify underlying genetic predispositions to depression and thus have not yet tested environmental, health, socio-behavioral or other non-genetic factors. Future analyses using RP to examine the impact of these factors on the development of depression would be anticipated to identify gene–non-genetic factor interactions. Indeed, the predictive gains of G × G analyses were stronger for men than for women, despite the fact that depression occurs disproportionately in women (∼2:1 female-to-male ratio21–25). This suggests that environmental factors may be needed in addition to genetic factors in understanding the aetiological pathways for women. Indeed, biological factors such as hormonal changes related to reproductive status26 ,27 may impact environmental factors such as psychosocial experiences (trauma, stress, interpersonal relationships, etc) and general health issues in the development of depression.
Genetic and biological correlates of depression
Numerous studies have identified SNPs that associate with depression. Many of the SNPs associated with depression from other studies were not significantly associated in our study. This is perhaps not surprising since a single factor is unlikely to provide consistent association especially in a complex condition such as depression, where multiple pathways intersect in regulating the risk of the disease. For example, if a SNP within the serotonin pathway also requires a SNP in the glutamatergic pathway in order for the patient to present with depression, the presence of either SNP in the absence of the other will not be predictive of depression. Moreover, as indicated by Shi and Weinberg,28 since the human genome contains genetic redundancy, disruption of a single gene may be selectively neutral, but the malfunction of several genes in a pathway might result in expression of a particular phenotype.
Both the primary splits in men and women were SNPs linked with DRD2 (dopamine receptor D2), a gene that has previously been linked with depression and social phobia.29–31 The primary male genotype split rs1800497, technically found in gene ANKK1, is historically known as the DRD2 Taq1A allele because of its known association with decreased dopamine receptor D2 density (in those with T-alleles).32–35 The Taq1A allele has also been previously associated with depressive symptoms in children, where those with the A1 allele (T) were more likely to have depressive symptoms.36 We saw a similar association between A1 and depression in WLS men, where those with two A1 alleles had 2.6 times higher odds for depression compared with those with one or no A1 alleles. The primary split in women (DRD2 rs2242592) has previously been found to be associated with schizophrenia, where the C-allele was associated with higher susceptibility for the disease.37 Interestingly, this same study also found the Taq1A allele to also associate with schizophrenia.
The secondary and tertiary right-hand splits in the female RP tree—FTO (fat mass and obesity associated) rs1421085 and IL6 (interleukin 6) rs1800795—have also been found to relate with mental illness and depression in previous studies.38 ,39 There is evidence that activin receptor signalling also is involved in affective disorders, especially when considering interaction with GABAergic pathways.40 Although we did not see an interaction between SNPs in GABA/activin receptor genes and depression, ACVR2B was associated with depression in women. No previous associations between depression and APOC3, ACVR2B, or GNRH1 have been reported.
That these genetic variants are associated with neuroendocrine pathways (GNRH1, ACVR2B) that are known to regulate neurotransmitter release and cognitive behaviour39 ,40 supports these associations as relevant to the aetiology of depression and underlines the benefits of using RP to identify meaningful G × G interactions associated with disease.
Limitations
Given the numerous genetic and non-genetic influences that are linked to depression and the small number of SNPs analysed, it is not surprising that predictability from our models was low (although our predictability was superior to previous studies examining only single main factors). Also, the predictive value of our statistical models was further limited due to user bias in selection of SNPs (from nearly 2 million SNPs in the human genome) used in this study. As a result of this, interactions we have found could potentially be moderated by another gene that we have not considered in this study. Nonetheless, we identified significant G × G interactions between known, and newly identified, loci associated with depression. Importantly, four of the seven SNPs identified in these interactions were primarily located in two pathways well known to impact depression: neurotransmitter and neuroendocrine signalling.
The results from the RP analyses conducted in this study were confirmed by LR, demonstrating the utility of RP as a screening tool for identifying meaningful G × G interactions. Future development of algorithms for RP analysis should maximise the distance between branches of the next best split (ie, rpart) and consider subsequent future split combinations that could potentially result in trees with ‘better’ overall predictability.
Summary
Our data indicate that G × G interaction analyses allow for enhanced predictability of conditions and diseases of ageing. RP is an efficient and powerful exploratory analysis technique for elucidating G × G interactions in large data sets and combined with LR provides an important statistical analysis for the identification of well-supported G × G interactions. We predict that such analytical methods will play an increasingly important role in the identification of epistatic effects in future GWAS. Finally, our studies illustrate how RP analyses can be used to find interacting pathways involved in the aetiology of a disease or condition such as depression.
Acknowledgments
This research uses data from the Wisconsin Longitudinal Study (WLS) of the University of Wisconsin-Madison. Since 1991, the WLS has been supported principally by the National Institute on Aging (AG09775, AG21079 and AG33285), with additional support from the Vilas Estate Trust, the National Science Foundation, the Spencer Foundation and the Graduate School of the University of Wisconsin-Madison. A public use file of data from the WLS is available from the Wisconsin Longitudinal Study, University of Wisconsin-Madison, 1180 Observatory Drive, Madison, Wisconsin 53706 and at http://www.ssc.wisc.edu/wlsresearch/data/. This material is the result of work supported with resources at the William S. Middleton Memorial Veterans Hospital, Madison, Wisconsin. The opinions expressed herein are those of the authors. The contents do not represent the views of the Department of Veterans Affairs or the US Government. This is Geriatrics Research, Education and Clinical Center VA paper # 2012–03.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplementary table 1
- Data supplement 2 - Online supplementary table 2
- Data supplement 3 - Online STROBE statement
Footnotes
To cite: Roetker NS, Yonker JA, Lee C, et al. Multigene interactions and the prediction of depression in the Wisconsin Longitudinal Study. BMJ Open 2012;2:e000944. doi:10.1136/bmjopen-2012-000944
Contributors CSA, RMH and TSH conceptualised the study. RMH, TSH, CLR, NSR, CL and CSA collected saliva samples and performed genotyping analyses. NSR, JAY, CL, VC and JJB performed statistical analyses on the Wisconsin Longitudinal Study data set. CSA and RMH directed the statistical analyses. NSR and CSA drafted the manuscript. All authors critically reviewed the manuscript and approved the final version.
Funding The study was supported by the National Institute on Aging (AG09775, AG21079 and AG33285).
Competing interests None.
Patient consent Obtained.
Ethics approval Ethics approval was provided by the Social Sciences Institutional Review Board, UW-Madison.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement WLS public release data is available for download at http://www.ssc.wisc.edu/wlsresearch. Information on obtaining WLS genotypic data is available at this site. All WLS data is available free of charge.