Article Text

Abstract
Introduction Chronic pelvic pain (CPP) affects >1 million UK women. Annual healthcare costs are estimated at >£150 million. Proven interventions for CPP are limited, and treatment is often unsatisfactory. Gabapentin is increasingly prescribed due to reports of effectiveness in other chronic pain conditions, but there are insufficient data supporting value in CPP specifically. The mechanism by which gabapentin exerts its analgesic action is unknown. Given the prevalence and costs of CPP, the authors believe that a large, multicentre, placebo-controlled, double-blind randomised controlled trial to evaluate the efficacy of gabapentin in management of CPP is required. The focus of this study is a pilot to inform planning of a future randomised controlled trial.
Methods and analysis The authors plan to perform a two-arm, parallel, randomised controlled pilot trial. The authors aim to recruit 60 women with CPP in NHS Lothian and NHS Grampian (UK) and randomise them to gabapentin or placebo. Response to treatment will be monitored by questionnaire compared at 0, 3 and 6 months. The primary objective is to assess recruitment and retention rates. The secondary objectives are to determine the effectiveness and acceptability to participants of the proposed methods of recruitment, randomisation, drug treatments and assessment tools and to perform a pretrial cost-effectiveness assessment of treatment with gabapentin.
Ethics and dissemination Ethical approval has been obtained from the Scotland A Research Ethics Committee (LREC 12/SS/0005). Data will be presented at international conferences and published in peer-reviewed journals.
Trial registration number ISRCTN70960777.
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
Article summary
Article focus
Is it possible to achieve acceptable recruitment and retention rates in a two-arm, parallel, randomised controlled pilot trial of women with CPP given gabapentin and placebo?
Are the proposed methods of recruitment, randomisation, drug treatments and assessment tools in this pilot trial effective and acceptable to participants?
Is gabapentin likely to be cost effective for the management of CPP in women given the current level of uncertainty?
Key messages
CPP affects >1 million UK women, and treatments are often unsatisfactory.
Gabapentin is being increasingly prescribed due to reports of effectiveness in other chronic pain conditions, but there are insufficient data supporting its value in CPP specifically.
A large, multicentre, placebo-controlled, double-blind randomised controlled trial (RCT) to evaluate the efficacy of gabapentin in management of CPP is required.
The focus of this study is a pilot to inform planning of a future RCT.
Strengths and limitations of this study
We recognise that there may be potential difficulties in mounting a large RCT for a chronic pain condition using gabapentin (a medication with known sedating side effects).
We have designed this pilot study to assess practical feasibility.
The study has been designed following the ‘Initiative on Methods, Measurement, and Pain Assessment in Clinical Trials’ recommendations for the design of chronic pain clinical trials.
Gabapentin requires titration to achieve an efficacious dose so that the rate and severity of adverse effects are minimised.
We have included focus group assessment of the acceptability of the drug treatment and titrating regime in our study to inform the design of the future RCT.
We plan to use a wide range of data collection tools in our study, but it is our intention to use fewer in our future RCT depending on their effectiveness in the pilot study (defined by lack of missing data, ability to detect effect and independence) and participant feedback on acceptability.
Introduction
Chronic pelvic pain (CPP) affects over 1 million women in the UK.1 2 It is the reason for 20% of gynaecological consultations and causes a 45% reduction in work productivity.3 The annual cost for caring for UK women with CPP has been estimated at £154 million. The cause of the painful symptoms experienced by women with CPP is poorly understood. Pain is often associated with specific pathological processes, such as endometriosis, but up to 55% of women with CPP appear to have no obvious underlying pathology.2 The management of CPP is difficult4 because in the absence of underlying pathology, no established gynaecological treatments are available.
Gabapentin (a GABA analogue) is being increasingly prescribed in general practice for CPP. It is also recommended by some practitioners as a treatment of choice for CPP in a multidisciplinary setting, despite no clinical evidence on which to base this recommendation. To our knowledge, only one study has evaluated the use of gabapentin for CPP. This small study (56 patients) compared gabapentin against amitriptyline for treatment of CPP and showed that gabapentin had greater efficacy (80% compared with 70% improvement in pain scores at 12 months).5 Unfortunately, this study had no placebo arm and the significance of the effect on quality of life provided by gabapentin in the management of CPP was not evaluated. Nevertheless, the efficacy of gabapentin has been documented for other chronic pain conditions: painful diabetic neuropathy, postherpetic neuralgia, mixed neuropathic pain conditions, spinal cord injury and phantom limb pain.6 The number needed to treat for improvement in all trials with evaluable data is 4.3 (95% CI 3.5 to 5.7). In some of these trials, gabapentin also improved sleep, mood and other elements of quality of life. The mechanism by which gabapentin exerts its analgesic action is unknown.
Ideally, a definitive evaluation of the efficacy of gabapentin in the management of CPP with no obvious underlying pathology requires a large, multicentre randomised controlled trial (RCT). This protocol outlines a pilot study to assess the processes that are vital to the delivery of such a trial.
Objectives
Primary objective
The primary objective is to determine whether it is possible to achieve acceptable recruitment and retention rates in two UK centres (NHS Lothian and NHS Grampian) within defined inclusion/exclusion criteria.
Secondary objectives
To determine the effectiveness and acceptability to patients of the proposed methods of recruitment, randomisation, drug treatments and assessment tools.
To determine whether gabapentin is likely to be cost effective given the current level of uncertainty and to ascertain what further evidence is needed for the evaluation of gabapentin.
End points
Primary end points
The proportion of eligible patients randomised into the study.
The proportion of randomised patients who take all their medication and fully complete questionnaires at final follow-up.
Secondary end points
Data on effectiveness and acceptability of proposed methods of recruitment, randomisation, drug treatments and assessment tools will be used to refine the design of the definitive RCT. The potential cost-effectiveness of gabapentin in the management of CPP will also be determined.
Methods and analysis
Study design
We aim to perform a two-arm, parallel, randomised controlled pilot trial (figure 1). This will be a two-centre study with recruiting in NHS Lothian (Edinburgh) and NHS Grampian (Aberdeen). We will recruit 60 patients over approximately 9 months. After randomisation, the participants will receive treatment for 6 months. Participants and the healthcare team will be unblinded at the end of their treatment.

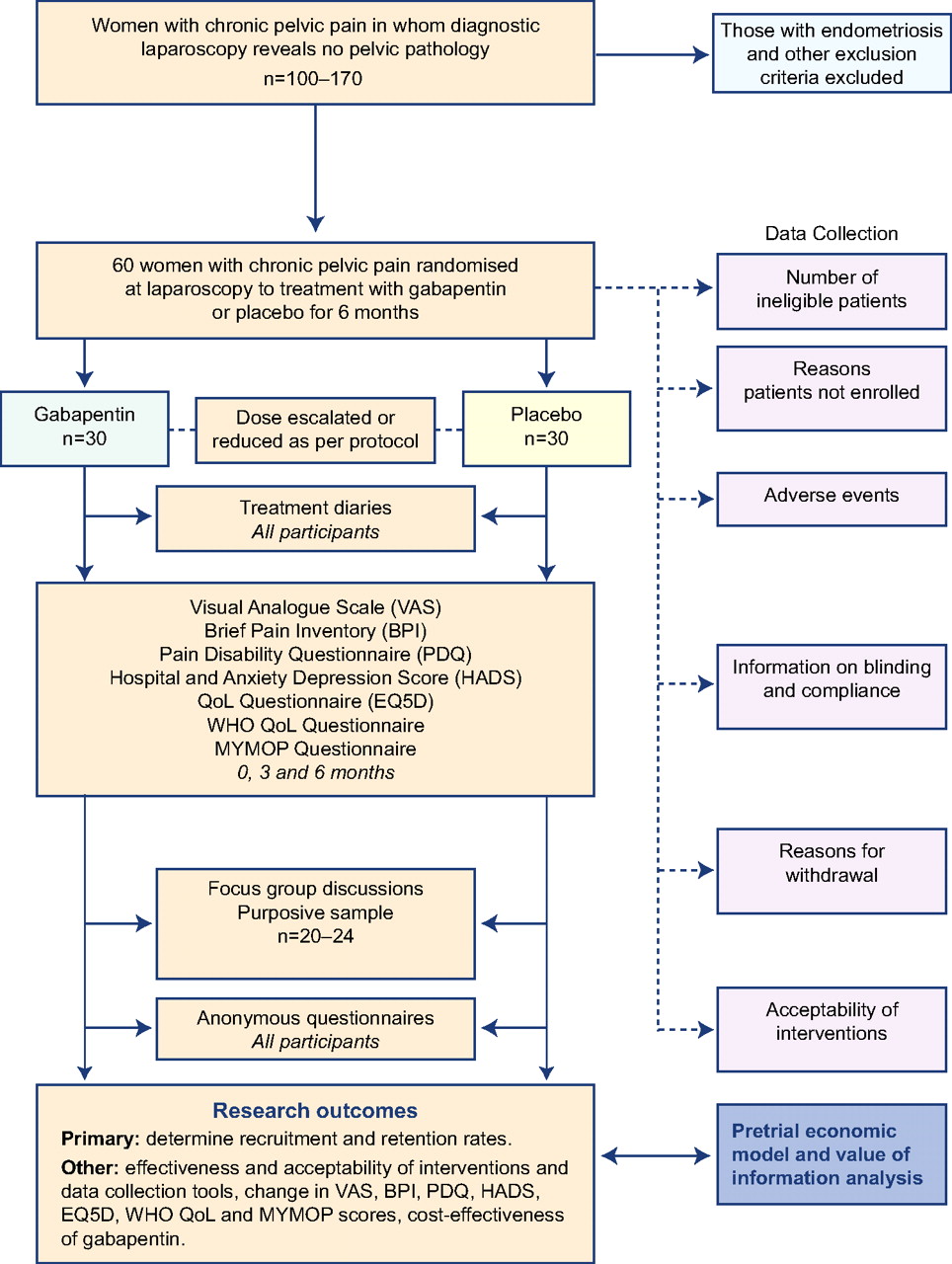{kind=link}
Flow of participants through the study. QoL, quality of life.
Subjects
A total of 60 women aged 18–50 years with a history of pelvic pain (cyclical or non-cyclical) and/or dyspareunia with no obvious pelvic pathology detected at laparoscopy will be invited to participate in the trial.
Study settings
We will recruit patients from gynaecology outpatient clinics, gynaecology wards and day surgery units within NHS Lothian and NHS Grampian.
Sample size
We have used a CI approach7 to estimate the sample size to establish feasibility based on a loss to follow-up of <20%. A 95% CI for 20% of 60 patients (12/60) is 11%–32%. We estimate that we will recruit approximately three to four patients per month from each centre and aim to recruit 60 patients over a 9-month recruitment period. Each centre performs six to seven laparoscopies per month that fit the inclusion criteria.
Inclusion criteria
Women aged between 18 and 50 years.
Pelvic pain of >6 months.
Pain located within the true pelvis or between and below anterior iliac crests, associated functional disability.
No obvious pelvic pathology at laparoscopy (<6 months and >2 weeks ago).
Using effective contraception.
Exclusion criteria
Known pelvic pathology, for example, endometriosis, cyst.
Taking gabapentin or pregabalin.
Due to undergo surgery in the next 6 months.
History of significant renal impairment.
Allergic to gabapentin.
Breast feeding.
Pregnancy or planning pregnancy in the next 6 months.
Participant enrolment
All gynaecology consultants within NHS Lothian and NHS Grampian will be sent a letter informing them of the study and requesting permission to approach their patients. Two research nurses (one in NHS Lothian and one in NHS Grampian) will be employed for the duration of the study to approach eligible women, provide them with patient information sheets and offer them the opportunity to discuss the trial, and obtain informed consent. Consent will only be taken once the patient has had ample time to read the patient information sheet and had her questions answered.
Intervention and randomisation
Eligible women will be randomised to either gabapentin or placebo using a web-based system. Women will be stratified by centre (NHS Lothian and NHS Grampian). We will use randomised blocks of varying sizes.
Dose regime
Participants will start on 300 mg gabapentin daily and will increase in 300 mg increments each week until they report a 50% pain reduction or side effects (eg, dizziness, somnolence, mood changes, appetite and poor concentration), up to a maximum dose of 2700 mg. Patients will be advised regarding their dosing regime weekly by a member of the research team who will phone until optimum dose is reached. It will be recommended that the drug should be taken in three equally divided doses daily. Participants will be advised to remain on the maximum tolerated dose for up to 6 months. The same protocol will be used for the placebo. When the participant stops treatment then the dose will be tapered down over 7–10 days at the clinician's discretion. Patients will be allowed to use other medication (including analgesics, self-medication and alternative treatments, eg, acupuncture) throughout the study period.
Data collection
Data storage
A log with the patients' name and date of birth will be kept along with their unique study number in a separate file. All the data generated from the study will be stored in an anonymised form in a bespoke database, which will also be password protected. Only anonymised information will be stored on this, and participants will only be identifiable by their study number. All paperwork will be kept in a locked filing cabinet in a locked office. All data will be stored on university server on a password-protected computer with limited access to the research team, in accordance with NHS and University of Edinburgh guidelines and in accordance with the Data Protection Act.
Screening
A member of the research team will carry out a screening visit to assess eligibility. All data will be recorded on a case record form and transferred to a secure database.
Participant log
The clinical research team will keep an electronic log of women who fulfil the eligibility criteria, women who are invited to participate in the study, women recruited and women who leave the trial early. Reasons for non-recruitment (eg, non-eligibility, refusal to participate, administrative error) will also be recorded. We will attempt to collect reasons for non-participation from women who decline to take part after previously providing contact details. During the course of the study, we will document reasons for withdrawal from the study and loss to follow-up. Participants will be reviewed by the clinical research team at 6 weeks, 3 months and 6 months.
Treatment diaries
All medications and healthcare resource use taken after screening and any medication other than the trial treatment taken during the study will be recorded in a treatment diary. This includes prescription and non-prescription treatment, such as contraceptives, vitamins, topical preparations, herbal preparations and non-pharmacological therapies.
Questionnaires
A questionnaire will be given to all participants at randomisation (0 months) and at 3 months. This will include the following validated tools:
Visual Analogue Scale (VAS).
Brief Pain Inventory.
Pain Disability Questionnaire.
Hospital Anxiety and Depression Score.
EQ5D QoL.
WHO QoL.
MYMOP patient-generated outcome questionnaire.
The questionnaire at 0 months will include questions to capture the baseline demographic and clinical characteristics of the participants.
A further questionnaire will also be given to all participants at 6 months, which will include the above and additional questions on whether they believed that they were receiving gabapentin or placebo and also questions on acceptability of the allocated medication/treatment regimes (and compliance) and on the acceptability of the above data collection methods. Lastly, we will ask the participants to complete a brief anonymous questionnaire once they have submitted their treatment diaries to assess level of adherence to diary keeping.
Focus groups
A purposive sample (based on age, social class and severity of symptoms) of 10 Edinburgh women (including some undergoing fMRI) and 10 Aberdeen women will be invited to participate in focus group discussions of the trial experience 6 months into the trial.8 Women who do not wish to participate in a focus group will be offered individual interviews using the same interview schedule. This will enable important issues arising in the focus groups to be explored in greater depth. Up to 20 interviews will be performed. Group/individual interviews will be audio-recorded, transcribed verbatim and analysed thematically to identify the issues of importance to participants not covered in the questionnaires, their feelings about trial participation and experiences with prescribed medication.
Healthcare resource utilisation measures
Information will be derived from treatment diaries and from research nurse reviews of the participants' hospital and general practitioners' records.
Adverse events
Participants will collect information about adverse events in their treatment diaries. However, they will be instructed to contact the clinical research team at any time after consenting to join the trial if they have an event that requires hospitalisation or an event that results in persistent or significant disability or incapacity. Gabapentin is generally well tolerated in the management of other chronic pain conditions, and serious adverse events are not anticipated. Any serious adverse events that occur after joining the trial will be reported in detail in the participant's medical notes, followed up until resolution of the event and reported to the ACCORD Research Governance (http://www.accord.ed.ac.uk) and QA Office based at the University of Edinburgh immediately or within 24 h.
Termination of study
Participants (and their gynaecologists) will be unblinded at the end of the study period (6 months). There will be no central unblinding facility, but the site pharmacies will be provided with the key that links drug pack number to treatment. Thus, it will be possible for unblinding (emergency or otherwise) to be carried out by a pharmacist if requested. All participants will be given the right to be unblinded, discontinue the drug or completely withdraw from the study at any time for any reason. Reasons for unblinding, before the termination of the study, will be collected. Those participants who feel that they have benefited from treatment with gabapentin, during the study period, will be advised to discuss continuation of treatment with their gynaecologist.
Proposed analyses
Determine recruitment and retention rates
Using the information collected from the participant log, we will determine the number of patients recruited from the pool of eligible women and a >50% recruitment will be deemed acceptable. While a retention rate of 100% would be ideal, we will consider a rate of 90% satisfactory. We will provide an estimate of the proportion and its 95% CI. If retention rates are low, we will use the information collected from the focus group discussions to ascertain why and improve compliance in the future trial. In addition, we will determine the nature and number of unanswered questions in each questionnaire and identify reasons for non-response through the focus groups and participant interviews in order to optimise data collection in the future trial.
Effectiveness and acceptability of proposed methods of recruitment, randomisation, drug treatments and assessment tools
These areas will be explored in the focus group discussions and assessed quantitatively using additional questions included in participant questionnaires administered at 6 months. Due to the conflicting literature about the benefits of methods such as prescription monitoring, pill counting and devices for monitoring the self-administration of medicines,9 data on blinding and compliance to treatment will be derived from questionnaires at 6 months. We aim to determine if treatment is acceptable in terms of self-reported compliance (from treatment diaries). Although this is a pilot study and the sample size is small, we will assess the effect of any non-compliance on the LICKERT score by performing protocol and intention-to-treat analyses. This information along with health professionals' and clinical research nurses' views (as assessed by questionnaire) will be used to inform the design of the future RCT. In addition, the difference in VAS scores between participants on gabapentin and placebo at 0 and 6 months will be assessed using analysis of covariance adjusting for baseline VAS score.
Pretrial economic model and value of information analysis
In addition to data relating to the clinical and quality-of-life parameters, data on healthcare resource use will also be collected. A decision model will be developed, from the perspective of the NHS, to estimate the costs and health outcomes in terms of quality of life and quality-adjusted life-years associated with gabapentin and placebo based on the data from this pilot study and the literature. A probabilistic decision model will be constructed to simulate the clinical pathways associated with gabapentin and placebo, according to the guidance set out by National Institute for Health and Clinical Excellence.10 The basic model structure will consist of two arms, replicating the clinical consequences of patients receiving gabapentin and placebo. The main data source relating to the key parameters of the model will be provided by the pilot study. The mean costs and quality-adjusted life-years associated with both arms will be calculated for the modelling period (duration of the trial). Cost–utility analysis will be carried out, and incremental cost per quality-adjusted life-years gained will be calculated. Particular consideration will be given to the potential for cost-effectiveness to vary by particular patient characteristics or risk groups where suggested by the literature. Probabilistic sensitivity analysis will be used to characterise uncertainty in parameters of the model and presented using cost-effectiveness acceptability curves. Standard univariate sensitivity analysis will be carried out to explore areas of structural uncertainty in the analysis. Finally, a value of information analysis on the expected value of perfect information will also be carried out to quantify potential value of further research based on the difference between expected net benefit with perfect information and existing information.
Ethics and dissemination
Ethical approval has been obtained from the Scotland A Research Ethics Committee (LREC 12/SS/0005). Data will be presented at international conferences and published in peer-reviewed journals. We will make the information obtained from the study available to the public through national bodies and charities.
Discussion
We believe that a definitive evaluation of the efficacy of gabapentin in the management of CPP requires a multicentre randomised placebo-controlled trial (RCT). Recognising that there may be potential difficulties in mounting a large RCT for a chronic pain condition using a medication with known sedating side effects and that requires a titrated dosing regime, we have designed this pilot study to assess practical feasibility following the IMMPACT (Initiative on Methods, Measurement, and Pain Assessment in Clinical Trials) recommendations for the design of chronic pain clinical trials.11 We are aware that our pilot study has a number of positive and negative aspects, and these are discussed below.
For our pilot study, we are using the most common design in confirmatory trials of chronic pain treatments: a ‘parallel groups’ design.11 We will randomise participants to either gabapentin or placebo and then evaluate recruitment and retention rates as our primary outcome. We appreciate that this design may be limited by the fact that the severity of the participants' pain may preclude them from remaining on the placebo for the 6-month follow-up period. Therefore, the outcome of the pilot will determine whether we need to consider alternative designs, such as ‘crossover’, ‘randomised withdrawal’ and ‘dose–response’ designs, for the future RCT assessing efficacy of gabapentin for CPP.
The criteria for inclusion and exclusion of study subjects into our pilot study are broad in attempt to reflect the real clinical scenario for prescribing gabapentin. The criteria do not take into account pain intensity, do not exclude women with non-reproductive comorbidities (eg, irritable bowel syndrome, interstitial cystitis) that could explain their symptoms and allow participants the use of concomitant medications. We are aware that these characteristics may increase variability in patient responsiveness to treatment and carry the risk of failing to demonstrate treatment effect. We will therefore capture this information in our pilot study in the participants' questionnaires and treatment diaries to inform interpretation of our results and the planning of the future RCT.
Like many of the medications used for chronic pain, gabapentin requires titration to achieve an efficacious dose so that the rate and severity of adverse effects are minimised. The duration of this titration period may be as many as 8–10 weeks. The 6-month follow-up in our pilot study allows for a 12-week maintenance phase that has become standard for confirmatory trials. It could be argued that a longer trial would be better to assess the long-term effects of gabapentin. On the other hand, we are aware that extended duration could be problematic because of the number of dropouts from the placebo arm due to inadequate pain relief. We believe that focus group assessment of the acceptability of the drug treatment and titrating regime in our pilot will therefore be essential in designing the future RCT.
The comparison of an investigational treatment with placebo is considered the gold standard for assessing efficacy and safety when a delay in the onset of treatment does not cause any lasting adverse effects and assuming that subjects fully understand their right to withdraw from the trial at any time for any reason.12 13 However, gabapentin is sedating and it can be argued that this increases the likelihood that both subjects and investigators can successfully guess to which group a subject has been allocated. We are therefore going to ask the subjects and investigators at the conclusion of the trial to guess the subjects' treatment group and the primary reason for the guess to determine whether significant ‘unblinding’ was present within the trial. This will determine whether we need to use an ‘active placebo’ mimicking the side effects of gabapentin in the future RCT.
Our data collection tools were chosen with advice from a clinical psychologist with a specialist interest in chronic pain and a general practitioner with a research interest in medically unexplained symptoms. The selection of these tools was also again based on the IMMPACT recommendations,11 that is, the need to assess the core domains of pain, physical/emotional functioning (including sleeping difficulties), improvement/satisfaction with treatment, symptoms and adverse events and participant disposition. We plan to use a wide range of data collection tools, but it is our intention to use fewer in our future RCT depending on their effectiveness in the pilot study (defined by lack of missing data, ability to detect effect and independence) and participant feedback on acceptability.
We also aim to determine whether gabapentin is expected to be cost effective given the current level of evidence and uncertainty through an iterative approach to economic evaluation of health technologies.14 15 Important gaps and uncertainty surrounding existing data and the expected cost-effectiveness will be explored through synthesis, modelling and value of information analysis prior to a definitive RCT. We will determine whether further evidence is needed to reduce the uncertainty surrounding cost-effectiveness, and if so, identify the focus of further research in terms of study design and data collection; this may have implications on determining an appropriate sample size (eg, powered to detect difference in clinical effect or cost-effectiveness).
Finally, although the primary outcome in our pilot study is to determine recruitment and retention rates, we will also measure change in VAS over 6 months. This combination of information will allow us to determine the effect size and SD and plan the sample size for the definitive RCT. Analyses of similar studies using gabapentin for chronic pain with VAS score as primary outcome indicate that the mean absolute difference in the VAS score comparing gabapentin against placebo ranges between 0.8 and 1.8, with an SD of approximately 2.5 after 1–2 months of treatment.6 Thus, our definitive RCT is likely to be powered to find a difference of >1.2 on the VAS scale (a clinically important symptom alleviation is defined as a reduction in VAS of >1.216 between the gabapentin and placebo arms of the study.)
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement 1
- Data supplement 10 - Online supplement 10
- Data supplement 2 - Online supplement 2
- Data supplement 3 - Online supplement 3
- Data supplement 4 - Online supplement 4
- Data supplement 5 - Online supplement 5
- Data supplement 6 - Online supplement 6
- Data supplement 7 - Online supplement 7
- Data supplement 8 - Online supplement 8
- Data supplement 9 - Online supplement 9
Footnotes
To cite: Horne AW, Critchley HOD, Doust A, et al. GaPP: a pilot randomised controlled trial of the efficacy of action of gabapentin for the management of chronic pelvic pain in women: study protocol. BMJ Open 2012;2:e001297. doi:10.1136/bmjopen-2012-001297
Contributors AH: research, contribution of original material, editing and approval of final manuscript; HC, AD, DF, JW and SJ: contribution of original material, editing and approval of final manuscript; OW, MP, SL and SB: research, contribution of original material, editing and approval of final manuscript.
Funding This work is supported by a grant from the Chief Scientist's Office Scotland (CZH/4/688). AH is funded by an MRC Clinician Scientist Fellowship (G0802808). The funders and study sponsor will have no role in the study design; collection, management, analysis and interpretation of data; writing of the report; and the decision to submit the report for publication.
Competing interests AH, HC, SJ, JW, SL, OW, MP and SB are all co-investigators on the grant from the Chief Scientist's Office Scotland (CZH/4/688) that has been secured to support this study. AH is funded by an MRC Clinician Scientist Fellowship (G0802808). HC holds an MRC Centre Grant (G1002033) and a project grant from Bayer Schering Pharma AG. AH and HC hold the University of Edinburgh Patent ‘Identification of Ectopic Pregnancies’ number 0712801.0.
Ethics approval Ethical approval has been obtained from the Scotland A Research Ethics Committee (LREC 12/SS/0005).
Provenance and peer review Not commissioned; internally peer reviewed.
Data sharing statement No additional data are available.