Article Text

Abstract
Objectives Control of the tuberculosis (TB) epidemic is a global health priority and one that is likely to be achieved only through vaccination. The critical overlap with the HIV epidemic requires any effective TB vaccine regimen to be safe in individuals who are infected with HIV. The objectives of this clinical trial were to evaluate the safety and immunogenicity of a leading candidate TB vaccine, MVA85A, in healthy, HIV-infected adults.
Design This was an open-label Phase I trial, performed in 20 healthy HIV-infected, antiretroviral-naïve subjects. Two different doses of MVA85A were each evaluated as a single immunisation in 10 subjects, with 24 weeks of follow-up. The safety of MVA85A was assessed by clinical and laboratory markers, including regular CD4 counts and HIV RNA load measurements. Vaccine immunogenicity was assessed by ex vivo interferon γ (IFN-γ) ELISpot assays and flow-cytometric analysis.
Results MVA85A was safe in subjects with HIV infection, with an adverse-event profile comparable with historical data from previous trials in HIV-uninfected subjects. There were no clinically significant vaccine-related changes in CD4 count or HIV RNA load in any subjects, and no evidence from qPCR analyses to indicate that MVA85A vaccination leads to widespread preferential infection of vaccine-induced CD4 T cell populations. Both doses of MVA85A induced an antigen-specific IFN-γ response that was durable for 24 weeks, although of a lesser magnitude compared with historical data from HIV-uninfected subjects. The functional quality of the vaccine-induced T cell response in HIV-infected subjects was remarkably comparable with that observed in healthy HIV-uninfected controls, but less durable.
Conclusion MVA85A is safe and immunogenic in healthy adults infected with HIV. Further safety and efficacy evaluation of this candidate vaccine in TB- and HIV-endemic areas is merited.
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
Article summary
Article focus
HIV infection increases susceptibility to TB, and globally, TB is the cause of death in up to half of AIDS deaths.
There is an urgent need for a safe and effective TB vaccine in HIV-infected people.
Key messages
MVA85A, a leading candidate TB vaccine, is safe and well tolerated in HIV-infected people and does not induce changes in either CD4 count or HIV RNA load.
MVA85A is immunogenic in HIV-infected people, and induces a similar immune profile to that seen in HIV-uninfected people, but the immunogenicity is less durable in HIV-infected people.
Strengths and limitations of this study
This is a Phase I study with 20 subjects, and further studies are needed in TB endemic countries in this important target population.
Introduction
Tuberculosis (TB) and HIV are inextricably linked. At the end of 2007, approximately 33.2 million persons were living with HIV-1 infection, an estimated one-third of whom were co-infected with Mycobacterium tuberculosis.1 TB is the cause of death for up to half of all AIDS patients,2 and the increasing incidence of drug-resistant strains of M tuberculosis poses a significant threat to a susceptible HIV-infected population.3
Mycobacterium bovis BCG fails to protect consistently against the adult pulmonary form of TB, while providing reliable protection against disseminated infection in childhood.4 An improved vaccine strategy is thus essential for global control of this disease.5 MVA85A (modified vaccinia virus Ankara expressing antigen 85A) is a leading candidate TB vaccine, designed to enhance the effect of BCG; it is safe and highly immunogenic in healthy BCG-naïve and BCG-vaccinated subjects, and in subjects latently infected with M tuberculosis in the UK and Africa.6–11
It is essential that any new TB vaccine is safe in an HIV-infected population. Subunit vaccines are an ideal choice for an immunocompromised population in which the safety of replicating whole organism vaccines may be a concern. Although MVA is a live viral vaccine vector, it cannot replicate in human cells.12 13 There are now safety data from a number of clinical trials with recombinant MVAs in HIV-infected subjects, which demonstrate no sustained effect on either HIV load or CD4 count.14–16 Most of these studies assessed immune-reconstituted HIV-infected individuals on antiretroviral therapy (ARV); however, some ARV-naïve subjects with more advanced HIV infection have also been vaccinated with a recombinant MVA, with no significant rise in HIV load or fall in CD4 count over a 4-week follow-up period.17 Preclinical studies in severely immunosuppressed macaques have also documented safety in this model.18 To date, MVA85A has been administered to more than 1000 individuals with no vaccine-related serious adverse events6 8–11 (McShane, unpublished data).
Here we present the first clinical trial of a subunit TB vaccine in an HIV-infected population. The primary endpoint was to evaluate the safety of two doses of MVA85A in healthy HIV-infected subjects in the UK, and the secondary endpoint was to evaluate the immunogenicity of this vaccine regimen.
Methods
Trial design and participants
The protocol for this multisite study was approved by the Medicines and Healthcare Products Regulatory Agency (MHRA), and ethical approval was obtained from the Gene Therapy Advisory Committee (GTAC). Participants were recruited from the Genitourinary Medicine (GUM) departments at the Oxford Radcliffe Hospitals NHS Trust, University Hospitals Birmingham NHS Foundation Trust (Selly Oak Hospital), Great Western Hospitals NHS Foundation Trust and Imperial College Healthcare NHS Trust, London.
Potentially eligible subjects were referred to the clinical trial team. Vaccination visits took place in Oxford; screening and follow-up visits took place at local NHS Trust sites. Written informed consent was obtained from all subjects prior to enrolment in the trial, and the referring HIV physicians were notified.
Eligibility required participants to be generally healthy, aged 18–55 years, with a normal chest radiograph and no clinical or radiological evidence of TB disease, a current CD4 count (measured at screening) of at least 350×106/l, a nadir CD4 count of not less than 300×106/l and an HIV RNA load at screening of less than 100 000 copies/ml. All subjects were diagnosed as having HIV infection at least 6 months before screening and had not received any ARVs within the preceding 6 months (full inclusion/exclusion criteria; table 1). In all cases, testing for latent M tuberculosis infection (LTBI) was conducted at screening using an in-house ex-vivo interferon γ (IFN-γ) ELISpot assay for ESAT6 and CFP10 peptides. Subjects found to be latently infected were referred back to their consultants for possible treatment at the end of the trial.
Full inclusion and exclusion criteria
The trial was registered on a clinical trials database: ClinicalTrials.gov ID NCT00395720.
Vaccine
Clinical grade MVA was produced under Good Manufacturing Practice standard by IDT Biologika GmbH (Dessau-Rosslau, Germany).
Procedures
The first group of 10 subjects were vaccinated intradermally with MVA85A at a dose of 5×107 plaque-forming units (pfu); the second group of 10 subjects were subsequently vaccinated with a dose of 1×108 pfu MVA85A. Subjects were sequentially allocated first to the low-dose group and then, once safety had been demonstrated, to the high-dose group. A diary card was completed by all subjects recording local and systemic adverse events and body temperature for days 1 to 7 postvaccination. All subjects were followed up regularly for 12 months, and blood samples were taken at each time-point for CD4 count, HIV RNA load measurement and cellular immunological assays. CD4 count and HIV RNA load results were communicated to the patient's HIV physician at regular intervals. Routine haematology and biochemistry assays were performed at weeks 1 and 12 after vaccination. All subjects attended their usual NHS follow-up appointments and received routine clinical care throughout the trial.
Immunological assays
Ex vivo IFN-γ ELISpot assays were performed at screening and at weeks 1, 2, 4, 12 and 24 postvaccination using fresh peripheral blood mononuclear cells (PBMCs), as described previously.10 A single pool of 15-mer peptides spanning the Ag85A protein (66, overlapping by 10 amino acids, 2 μg/ml final concentration of each peptide in each well) and seven pools of non-overlapping 9–10 Ag85A peptides (final concentration of 10 μg/ml in each well) were used to stimulate for the IFN-γ ELISpot assays. For the seven peptide pool wells, the results were summed across all peptide pools for each time-point. This potentially resulted in duplicate counting of T cells that responded to any of the 10mer overlap regions, since any 10mer occurred in two pools with adjacent peptides, but allows direct comparison with immunogenicity data from previous trials.
Intracellular cytokine staining of PBMCs was performed as described previously.19 Briefly, cells were stained with the amine reactive LIVE/DEAD fixable dye ViViD (Molecular Probes, Invitrogen, Eugene, Oregon) and then surface-stained with monoclonal antibodies against CD4, CD14, CD19, CD27, CD45RO, CD57 and CCR5. After washing, cells were permeabilised (Cytofix/cytoperm kit; BD Pharmingen, San Diego, California) and stained intracellularly for CD3, CD8, IFN-γ, TNF-α, IL-2 and MIP-1β. Naïve CD4 memory cells were identified as CD45RO-CD27+CD57+; memory CD4 T cell as CD45RO+CD27±CD57±. CD4 T cells producing two or more cytokines (IFN-γ, IL-2 or TNF-α) following stimulation with Ag85A peptides were sorted to greater than 98% purity using a modifed FACSAria flow cytometer (BD Biosciences, San Jose, California); CMV-specific CD4 T cells, defined similarly, and bulk resting memory CD4 T cells were sorted simultaneously. Cytokine production and the expression of surface markers were analysed using FlowJo Tree Star Inc version 8.7, Pestle version 1.5 and SPICE version 4.1 (both from Mario Roederer, Vaccine Research Center, National Institute of Allergy and Infectious Diseases, National Institutes of Health).
Real-time quantitative PCR (qPCR) was used to determine the proportion of HIV-infected Ag85A-specific CD4 T cells; resting memory CD4 T cells (non-Ag85A-specific) and CMV-specific CD4 T cells were used as intraindividual controls. HIV-1 gag proviral DNA within sorted CD4 T cell populations was quantified by qPCR, as described previously.20 qPCR was performed simultaneously for albumin gene copy number in all sorted CD4 T cell populations to quantify cell number in each reaction. HIV-1 gag primers for qPCR: the Gag clade B primer position and sequence were 795gagB-F: ggtgcgagagcgtcagtattaag, 911gagB-R: agctccctgcttgcccata, and the probe was 841gagB-P: FAM-aaaattcggttaaggccagggggaagaa-QSY7 (MegaBases). The Gag clade C primer position and sequence were 689gagC-F: ggggaagtgaYatagcagga, 841gagC-R: ggYccttgtYttatgtccaa, and the probe was 710gagC-P: ctactagtaVccttcaRgaacaRataRcatggatga. The albumin primer sequences were Alb-F: tgcatgagaaaacgccagtaa, Alb-R: atggtcgcctgttcaccaa, and the probe: Alb-P: FAM-tgacagagtcaccaaatgctgcacagaa-QSY7. The degenerate HIV gag clade A/G primer position and sequence were 783gagA/G-F: 5′-gagagagatgggtgcgagagcgtc-3′ (GAGAGAGATGGGTGCGAGAGCGTC), 895gagA/G-R: 5′-ctttccagctccctgcttgccca-3′, and the probe was 844gagA/G-P: 5′-attcggttaaggccagggggaaagaaaaaat-3′.
CD4 T cells from 12 subjects (4/10 in the low-dose group and 8/10 in the high-dose group) were analysed for HIV gag content; low sort yields (less than 250 antigen-specific CD4 T cells) prevented similar analyses in the remaining eight subjects.
Serum levels of MIP-1α, MIP-1β, RANTES, TNF-α and IL-2 were measured prevaccination and at weeks 1, 2 and 24 postvaccination using FlowCytomix simplex kits (Bender MedSystems, Vienna). Serum was thawed and plated out in duplicate in round-bottom 96-well plates. Samples were used neat for MIP-1α, MIP-1β, TNF-α and IL-2, and diluted 1/10 for RANTES. Standards (provided) were reconstituted as specified, and 1/3 serial dilutions plated in duplicate. Bead and biotin-conjugate cocktails were added to each well in 25 μl and 50 μl volumes, respectively, and incubated for 2 h in the dark at room temperature. Samples were then washed twice in 100 μl assay buffer. Streptavidin-PE (50 μl) and assay buffer (100 μl) were added, and plates were incubated for 1 h in the dark at room temperature. Samples were then washed again and transferred to cluster tubes in 4% paraformaldehyde in PBS (400 μl) and acquired using an LSRII flow cytometer (BD Biosciences) set up with the beads provided and the highest concentration standard; the stopping gate was set so that at least 300 events were collected for each sample. Data were analysed using FlowCytomixPro software (Bender MedSystems).
Statistical analysis
Continuous outcomes were presented as a median (range) for each group. The median difference was calculated to assess the difference between two groups, and the corresponding 95% CI was computed using the robust method proposed by Newson.21 The overall magnitude of vaccine-induced CD4 T cell responses over the 24 weeks was summarised using the area under the curve (AUC) for each dose group. Peak (1 week) and plateau (24 weeks) CD4 T cell responses were compared with those at baseline (prevaccination) using the Wilcoxon signed rank test, Stata Statistical Software, Release 9.0. 2005. The Mann–Whitney U test was used to compare between the vaccine groups. Comparisons between specific time points in this trial and previously published trials of MVA85A,6 22 and between low- and high-dose groups were conducted using the Mann–Whitney U test (Stata).
Non-parametric Spearman rank analyses were used to correlate immune assays and CD4/HIV RNA load with CD4 T cell responses.
Results
Thirty-five individuals attended for screening between October 2006 and June 2009; 20 subjects were enrolled (supplement figure 1, flow chart). The demographics of the 20 participants are shown in table 2 and were comparable across the two groups.
Subject demographics: comparison of low and high dose vaccination groups
Safety of MVA85A in HIV-infected subjects
Local reactions related to the MVA vaccine occur during the first week after vaccination and have been reported previously.6 7 10 The local adverse event (AE) profile recorded in the diary cards of the HIV-infected subjects in this study was comparable with that observed in HIV-uninfected subjects6 7 10 (Pathan et al, unpublished data), although pruritus appeared slightly higher in the HIV-infected group (table 3a). There were no severe local reactions, and most local AEs were mild.
One subject in the low-dose group experienced a single episode of fever greater than 37.5°C (documented at 38.1°C). All other subjective systemic symptoms occurred with similar frequencies in the low-dose and high-dose groups (table 3b). There was one moderate systemic AE, and all others were mild.
The prevaccination CD4 counts and HIV RNA loads for all 20 subjects over a time period of 1–6 years before enrolment are shown in figure 1. These longitudinal data illustrate the levels of natural fluctuation that occur over time and represent important baseline measurements for the interpretation of these parameters postvaccination, also shown in figure 1, in the absence of an unvaccinated control group. No clinically significant changes in CD4 counts or HIV RNA load were seen postvaccination.

(A–D) Comparison of pre- and postvaccination CD4 counts: longitudinal CD4 counts of subjects in the low-dose (5×107 pfu MVA85A) group pre-vaccination, (A); and high-dose (1×108 pfu MVA85A) group (C) prevaccination. Longitudinal CD4 counts postvaccination (up to 24 weeks) in the low-dose (B) and high-dose (D) groups. The x-axis for the prevaccination data is not standardised for time but varies from 1 to 6 years, depending on the individual subject. (E–H) Comparison of pre- and postvaccination HIV RNA loads: longitudinal HIV RNA loads of subjects in the low-dose (E) and high-dose (G) groups prevaccination. Longitudinal HIV RNA loads postvaccination (up to 24 weeks) in the low-dose (F) and high-dose (H) groups. Anonymous number codes (001–036) are shown in the key. The x-axis for the prevaccination data is not standardised for time but varies from 1 to 6 years depending on the individual subject.
A major concern with respect to the vaccination of HIV-infected individuals is the possibility that responding antigen-specific CD4 T cells will act as preferential targets for HIV infection as a function of their activated status, thereby enhancing the propagation of HIV and accelerating disease progression. In this study, however, there was no evidence for preferential HIV infection of Ag85A-specific CD4 T cells. The median number of CD4 T cells in Ag85A-specific qPCR reactions was 73 (95% CI 15 to 130 CD4 T cells). Where possible, multiple time-points throughout the vaccination course for each subject were analysed, to determine the proportion of Ag85A-specific CD4 T cells that were infected with HIV. Of 29 Ag85A-specific CD4 T cell samples in total (11 subjects), only one subject showed a positive signal for HIV gag DNA by qPCR at two different postvaccination timepoints (weeks 2 and 8 postvaccination; data not shown). In the 11 subjects tested, resting HIV-specific and CMV-specific memory cell populations showed a positive signal in all assays (data not shown). In addition, vaccination had no effect on levels of MIP-1α, MIP-1β, RANTES, TNF-α and IL-2 in unstimulated serum (data not shown).
Immunogenicity of MVA85A in HIV-infected subjects
MVA85A induced significant antigen-specific T cell responses as measured by the IFN-γ ELISpot assay (figure 2A–D, table 4). In the low-dose group, the frequency of IFN-γ-secreting T cells responding to the single 85A peptide pool at week 1 postvaccination increased by a median of 393 spot forming cells (sfc)/million compared with prevaccination baseline measured on the day of screening (p=0.009); there, a significant increase remained until 24 weeks postvaccination (p=0.032). In the high-dose group, a median increase of 502 sfc/million was apparent at week 1 postvaccination (p=0.005); again, this response was maintained until 24 weeks (p=0.048). In both groups, T cell responses to the summed 85A peptide pools followed a similar pattern with a significant increase above baseline at week 1 (738 and 1730 sfc/million for low- and high-dose groups, respectively), although, the week 24 response was only maintained significantly above baseline in the high-dose group (low dose p=0.17; high dose p=0.007). There were no significant differences in the overall magnitude (ie, AUC) of the IFN-γ response between the low- and high-dose groups across the follow-up period (p=0.29 and p=0.68 for summed and single peptide pools, respectively; data not shown). However, the responder rate was higher in the high-dose group with no observed non-responders (ie, those subjects with no measurable vaccine induced immune response) compared with 1–2 non-responders (depending on antigen) in the low-dose group. Overall, the kinetics of the Ag85A-specific T cell responses were very similar to those previously reported in similar studies of HIV-uninfected subjects.6–11

(A–D) IFN-γ ELISpot responses in the low-dose and high-dose groups. Longitudinal responses to the single 85A peptide pool are shown for the low-dose (5×107 pfu MVA85A) group, (A) and high-dose (1×108 pfu MVA85A) group (B). Longitudinal responses to the summed 85A peptide pools are shown for the low-dose (C) and high-dose (D) groups. Horizontal bars represent the median response. Comparison of IFN-γ ELISpot responses in HIV-infected and HIV-uninfected subjects (E–G). Responses to summed 85A peptide pools for low-dose and high-dose vaccine regimes at screening (E); week 1 postvaccination (F) and week 24 postvaccination (G). Subjects with LTBI are denoted by grey symbols. Statistically significant differences in the responses between HIV-infected and HIV-uninfected subjects were assessed using the Mann–Whitney U test. *p<0.05, **p<0.01, ***p<0.001. Horizontal bars represent the median response6 7 10 (Pathan et al, unpublished data).
Ex vivo IFN-γ ELISpot statistics (1): comparison of screening, week 1 and week 24 responses within low- and high-dose groups, for both summed and single pooled peptides, using the Wilcoxon signed rank test
Ag85A-specific T cell responses in the low-dose group were significantly lower compared with those observed in previously published studies of HIV-uninfected subjects (AUC analysis, p=0.02; table 5).6 22 This difference was not significant at week 1 (MWU analysis, p=0.12), which represents the peak response, but was significant at week 24 postvaccination (MWU analysis, p=0.004; table 5, figure 2E–G). There were no significant differences in baseline (prevaccination) responses between the HIV-infected and HIV-uninfected groups (MWU analysis, p=0.85; figure 2E). Similarly, HIV-infected subjects in the high-dose group exhibited significantly lower responses compared with HIV-uninfected subjects who received the same dose of MVA85A (AUC analysis, p=0.0001; week 1 and week 24 MWU analysis, p=0.0001; table 5, figure 3E–G). Again, baseline responses were comparable (p=0.24) (figure 2E).
Ex vivo IFN-γ ELISpot statistics (2): comparison of responses between HIV-infected and HIV-uninfected subjects, at each time-point and each vaccine dose, using Mann–Whitney U test

Ag85A-specific cytokine/chemokine production by CD4 T cells pre and post vaccination. MVA85A induces polyfunctional Ag85A-specific CD4 T cells in HIV-infected individuals. MVA85A vaccination-induced production of IFN-γ, IL-2, MIP-1β and TNF-α by antigen-specific CD4 T cells was assessed following Ag85A peptide stimulation of cryopreserved PBMC using polychromatic flow cytometry. (A) Individual data points shown with a median line, IQR (open bars) and range (whiskers) at baseline and at each time-point postvaccination for every possible combination of cytokine/chemokine production. High-dose and low-dose groups were analysed together (n=17). (B) Functional profile of the Ag85A-specific CD4 T cell response summarised in pie charts (n=17). CD4 T cells producing a given number of cytokines/chemokines are grouped and colour-coded together. Pie charts are shown for the high-dose HIV-infected group (n=9), low-dose HIV-infected group (n=8) and low-dose healthy controls (n=6). (C) Absolute percentages of the highest frequency CD4 T cell subsets producing specific combinations of chemokine/cytokines at week 1 postvaccination across the different groups; *p<0.05, **p<0.01.
There was no significant correlation between the CD4 count at screening and the peak summed 85A peptide pool response (R=0.04, p=0.09), nor between HIV RNA load at screening and the peak immune response (R=−0.04, p=0.08). There was a strong negative correlation between viral load at screening and the plateau immune response at week 24 (R=−0.8, p=0.002).
Four subjects were found to have LTBI. However, there were no significant postvaccination changes in the magnitude of ESAT-6/CFP10 responses in these subjects (data not shown).
Ag85A-specific cytokine/chemokine production by CD4 T cells prior to MVA85A vaccination was undetectable and increased significantly postvaccination (figure 3A). One week following MVA85A vaccination, more than 40% of Ag85A-specific CD4 T cells produced IFN-γ, IL-2, MIP-1β and TNF-α simultaneously (p<0.01); this remained significant at week 2 (p<0.05). By week 8 postvaccination, 60% of Ag85A-specific CD4 T cells were still producing IFN-γ, IL-2 and TNF-α simultaneously; production of IL-2 and IFN-γ was maintained until 24 weeks. Production of MIP-1β was not detected beyond week 2 postvaccination (figures 3A, 4A). In the first 2 weeks postvaccination, Ag85A-specific CD4 T cells exhibited similar functional profiles in the HIV-infected groups, both low-dose and high-dose, and the ‘control’ low-dose HIV-uninfected group; in all cases, the responses were dominated by four functionally distinct subsets (figure 3B,C). IFN-γ production measured by ICS correlated closely with IFN-γ secretion observed in the ELISpot assay (R=0.75, p<0.0001, n=45; data not shown).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
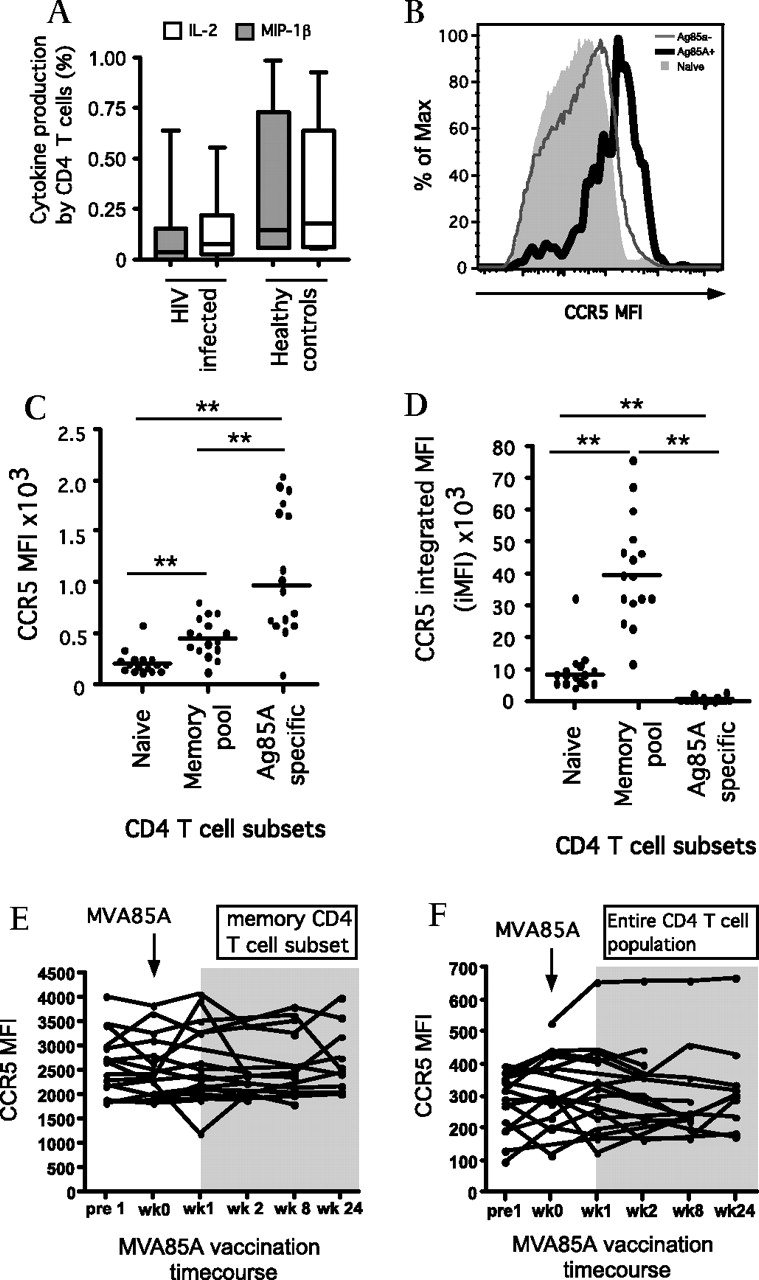
Cytokine/chemokine production and receptor expression. (A) Absolute percentage of Ag85A-specific CD4 T cells producing IL-2 or MIP-1β at week 1 post-MVA85A vaccination in HIV-infected subjects (low-dose and high-dose, n=17) versus healthy controls (n=6). (B) Representative histogram and (C) scatter plot showing CCR5 median fluorescence intensity (MFI) in naïve CD4 T cells, memory CD4 T cells and activated Ag85A-specific CD4 T cells at week 1 post vaccination (n=16 in all groups). (D) Scatter plot showing CCR5 integrated MFI (iMFI) for naïve, memory and Ag85A-specific CD4 T cells (n=16). Line charts showing CCR5 MFI in memory CD4 T cells (E) and the total CD4 T cell population (F) across the MVA85A vaccination time course (n=16). *p<0.05, **p<0.01.
Surface expression of the HIV M-tropic co-receptor CCR5 was monitored throughout the course of vaccination (figure 4). At week 1 postvaccination, the MFI for CCR5 was highest in the activated Ag85A-specific CD4 T cells and lowest in the naïve CD4 T cell subsets. Using the integrated MFI (iMFI) function calculated using both MFI and cell frequencies, expression of CCR5 was highest in the much larger memory CD4 T cell pool (figure 4D). No significant increase in CCR5 expression was detected in either the memory (figure 4E) or the total (figure 4F) CD4 T cell populations as a result of MVA85A vaccination.
Discussion
This is the first Phase I trial of a subunit vectored TB vaccine in HIV-infected individuals. The two main findings of this study are that MVA85A is safe in healthy subjects infected with HIV (a proportion of whom are co-infected with M tuberculosis), and that MVA85A is immunogenic in this population, albeit at a lower level than in HIV-uninfected subjects. As a result of these promising findings, similar studies in South Africa (Scriba et al, submitted) and Senegal (Mboup et al, unpublished data) are ongoing.
The local and systemic AE profiles in this trial were comparable with those observed in HIV-uninfected subjects previously. While we have not evaluated the safety of this vaccine in HIV-infected subjects with lower CD4 counts, we would not expect the safety profile of this vaccine to be altered by progression of HIV disease. Importantly, there were no clinically significant effects of vaccination on either CD4 count or HIV RNA load. It is important to understand the variability in these parameters during stable untreated HIV infection, to enable meaningful interpretation of fluctuations throughout the trial period. A recent quantitative review of cohort studies of HIV-infected adults suggests that, during stable untreated infection, intraindividual HIV RNA measurements can fluctuate over short periods of time by as much as 0.2–0.5 log10 copies/ml, with measurement error and physiological fluctuation each contributing about 50% of this variation. CD4 counts can also vary within patients by 60–130 cells/μl within weeks, but there is less measurement error, and during stable infection the overall within-subject variation is proportionally larger for HIV RNA than for the CD4 count.23 In this study, we also observed greater intraindividual variations in HIV RNA load compared with CD4 counts. Only one subject in each group demonstrated a more than 0.5 log rise in HIV RNA load at week 24 postvaccination. The first of these (low-dose group, 008) had displayed a relatively high baseline HIV RNA load and low baseline CD4 count at screening, just within the inclusion criteria. As the rise in HIV RNA load was only detectable 24 weeks after vaccination, it was considered highly unlikely that this was due to MVA85A vaccination. This subject commenced ARVs before week 24; all other subjects remained ARV-free throughout the trial period. The second subject (high-dose group, 035) developed a rise in HIV RNA load between week 12 and week 24, peaking at just greater than a 0.5 log increase over baseline. However, this subject's HIV RNA load had been stable prior to this, so it was again deemed unlikely that this was a vaccination-related effect. Other fluctuations in the HIV RNA loads were observed, but any sustained rise was well within the expected 0.5 log variation.
HIV preferentially infects memory CD4 T cells,24 25 in particular HIV-specific memory CD4 T cells,20 and other activated antigen-specific CD4 T cells.26 Using a sensitive qPCR method, we detected HIV gag DNA in Ag85A-specific CD4 T cells from only 1/11 subjects postvaccination (three low-dose and eight high-dose). This subject received high-dose MVA85A vaccination. This low positivity rate concurs with the stable CD4 count and HIV RNA load parameters in most subjects throughout the trial. Although the qPCR assay is sensitive, the low yield of Ag85A-specific CD4 T cells entering the assay is an important limitation and provides just a snapshot of the HIV burden within the Ag85A-specific CD4 T cell pool, and warrants further future investigation. However, while small effects of MVA85A on HIV infection of CD4 target cells cannot be excluded given the small sample size, these data suggest that MVA85A vaccination of healthy HIV-infected individuals does not lead to widespread preferential infection and depletion of vaccine-induced CD4 T cell populations in the periphery. These data are supported by no change in surface expression of the HIV co-receptor CCR5 following MVA85A vaccination. In addition, the lack of effect of vaccination on chemokine and cytokine levels in unstimulated serum supports the interpretation that vaccination with MVA85A did not lead to widespread immune activation in this subject group.
We have shown that vaccine-induced immunogenicity, while of moderate magnitude, is significantly lower than in healthy HIV-uninfected subjects given the same dose of MVA85A. Thus, additional strategies may be required to augment immune responses to MVA85A vaccination in the context of HIV infection. Partial reconstitution of the immune system with ARVs may improve the response to vaccination. Indeed, this study shows that the baseline HIV RNA level is a strong negative predictor of summed 85A peptide pool responses at week 24 postvaccination. In addition, a booster vaccination with MVA85A may be required to enhance the vaccine-induced immune response.
The functional profile of Ag85A-specific CD4 T cells observed in this study was remarkably comparable with that of healthy HIV-uninfected subjects19 and M tuberculosis-infected subjects,10 although the response in HIV-infected subjects is less durable. IFN-γ and TNF-α are known to be important for protective immunity to TB, and these cytokines dominated the MVA85A vaccine-induced CD4 T cell response elicited in the current study cohort. In this study, production of MIP-1β by Ag85A-specific CD4 T cells was not detectable after week 2 postvaccination. The production of multiple cytokines including IL-2 and MIP-1β by HIV-specific CD8 T cells is associated with long-term non-progression, albeit with uncertain causality.27 The production of β-chemokines by CD8 T cells was not assessed here, as there were no detectable Ag-85A specific CD8 T cell responses pre- or postvaccination.
Vaccine-induced bystander activation of CD4 T cells in HIV-infected subjects is likely to be undesirable because activated CD4 T cells are at increased risk of HIV infection. The overall pattern of cytokine production and the frequency of CMV-specific CD4 T cell producing cytokines remained both relatively stable over the 24-week period following MVA85A vaccination and comparable with prevaccination responses (supplemental figure 1). These data suggest that MVA85A vaccination does not induce substantial bystander activation of antigen-specific CD4 T cell populations.
Demonstrating the safety and immunogenicity of a new TB vaccine in an important target population is a crucial step but does not mean that the vaccine will be effective. However, the data presented here support further safety and immunogenicity studies of this candidate vaccine, together with proof-of-concept efficacy trials, in TB and HIV endemic areas.
Acknowledgments
We thank J Scott and the clinical research nurses in Oxford, at St Mary's Hospital in London and at Selly Oak Hospital in Birmingham, especially K Legg and J Harding, for their invaluable assistance in identification, recruitment and follow-up of eligible subjects. We also thank A Hill for discussions. Oxford University was the sponsor for this clinical trial.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data Supplement - Manuscript file of format pdf
Footnotes
To cite: Minassian AM, Rowland R, Beveridge NER, et al. A Phase I study evaluating the safety and immunogenicity of MVA85A, a candidate TB vaccine, in HIV-infected adults. BMJ Open 2011;1:e000223. doi:10.1136/bmjopen-2011-000223
Funding This study was supported by The Wellcome Trust (grant no WT076943MA), TBVAC (an EU 6th Framework programme grant) and The National Institute of Health Research Oxford Biomedical Research Centre.
Competing interests AAP and HMcS are named inventors on a composition of matter patent for MVA85A owned by the University of Oxford, and are shareholders in a Joint Venture formed for the further development of this vaccine.
Patient consent Obtained.
Ethics approval Ethics approval was provided by the Gene Therapy Advisory Committee.
Contributors Concept and design of study, and obtaining funding: HM. Protocol writing: HM, CRS, AMM, AML. Recruitment, vaccination and follow-up of volunteers: AMM, RR, IDP, MH, AW, JR, JS, GR. Immunology assays and analysis: AAP, NERB, IS, SH, HAF, HP. Collaboration and contribution of reagents/materials/analysis tools: DRA, DAP, JPC, DCD, RAK, MR. Data analysis: AMM, RR, AAP, NA, NERB, HM. Writing of the paper: AMM, HM, with contributions from all authors.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement No additional data available.