Article Text

Abstract
Background Medical devices are used widely for virtually every disease and condition. Although devices are subject to regulation, the number of recalls, the clinical data requirements for regulation and the impact on patient safety are poorly understood.
Methods The authors defined a device using European directives and used publicly available information on the Medicines and Health Regulatory Authority website to determine the number of devices recalled from January 2006 to December 2010. Two reviewers independently assessed Field Safety Notices and Medical Device Alerts. The authors wrote to manufacturers to obtain further information and clinical data, and summarised data by year, Conformité Européenne classification, indication, and Food and Drug Administration recall system of severity.
Results In total, 2124 field safety notices were issued over the 5-year period, an increase of 1220% (62 in 2006 to 757 in 2010). 447 Medical Device Alerts were issued in the same period, and 44% were assessed as a reasonable probability of causing serious adverse health consequences or death. The authors wrote to 192 manufacturers of withdrawn devices and received 101 (53%) replies; only four (2.1%) provided the clinical data the authors requested. A lack of available transparent data prevented full analyses of the safety impact. Of the highest-risk recalled devices, more than half were related to the cardiovascular system (25%) or musculoskeletal system (33%), and 88% (95% CI 80% to 97%) were assessed as a reasonable probability of causing serious adverse health consequences or death. For low-risk devices, the figure was 34% (95% CI 26% to 42%).
Conclusion The number of medical devices subject to recalls or warnings in the UK has risen dramatically. A substantial number of these devices may have caused serious adverse effects in patients and contributed to healthcare costs. Significant problems exist in the UK with a lack of access to transparent data and a registry of the highest-risk devices.
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
Article Summary
Article focus
To describe the number of medical-device recalls in the UK that occurred over a 5-year period from 2006 to 2010.
To determine the clinical data required at the time of regulation and the data available at the time of device recall.
To determine the potential risk to patients associated with recalled medical devices.
Key messages
There was a substantial increase in field safety notices over the 5-year period.
A substantial number of devices may have caused serious adverse effects in patients and contributed to healthcare costs over this time, but a lack of available transparent clinical data currently prevents full analyses of the safety impact of recalled devices in the UK.
Of the highest-risk devices recalled, more than half were related to the cardiovascular system or the musculoskeletal system.
Strengths and limitations of this study
Strengths
Quantification of all reported device recalls in the UK over a 5-year period.
A breakdown by Conformité Européenne marked classification of device.
An assessment of the potential harms of device recalls based on freely available published data on the Medicines and Health Regulatory Authority website.
Limitations
We were limited by a lack of available clinical data on recalled devices and the absence of a central registry, particularly of the highest-risk devices, which limited our ability to fully quantify and assess the implications of recalls on patient safety.
Our classification of devices and Food and Drug Administration recall status was based on two general practitioners clinical experience, which means they may differ from manufacturers' classification and other clinicians.
Owing to a lack of clinical data made available to us, we were unable to determine the reason for the rise in field safety notices. Also, we do not know when the problem first arose, and what kind of premarket clinical testing had been undertaken for many recalled devices.
Introduction
Medical devices are used for the diagnosis, monitoring and treatment of virtually every disease or condition, and include familiar objects such as simple bandages to high-end MRI scanners. Estimates suggest there is a vast array of devices in circulation, with some 500 000 medical devices worldwide available to healthcare providers and patients.1
Because of their vital role in healthcare, medical devices require regulatory approval. In Europe, they are subject to council directives of the European Union (EU) which stipulate that ‘devices must be designed and manufactured in such a way that, when used under the conditions and for the purposes intended, they will not compromise the clinical condition or the safety of patients.’2–4 These directives require medical-device manufacturers to display Conformité Européenne (CE) marking on their products as a way of ensuring/signifying that devices are safe and fit for their intended purpose. CE marking is conducted by EU-accredited private organisations called notifying bodies, rather than by a centralised regulator,5 who are responsible for the evaluation of the submitted clinical data by manufacturers.6 Each EU member state is responsible for overseeing this legislation. In the UK, it is the Medicines and Health Regulatory Authority (MHRA), which implements the European medical-device directives. However, regulation of medical devices has lagged behind that of drugs: formal regulation in Europe began only in the mid-1990s.7
Approval of medical devices is coordinated by the MHRA, which certifies notified bodies, which are for-profit organisations authorised to grant a CE-mark certification. Currently, in the European Union, there are 74 separate notifying bodies authorised in 25 countries to approve medical devices (six in the UK). Under the current system, a manufacturer selects a notified body to undertake certification of a new device for CE marking. The notified body will request certain materials (eg, a literature review), depending on the device class, and assess the manufacturer's conformity to the essential requirements listed in each directive. A CE mark for a medical device awarded in one country enables access to the entire EU market.
Device regulation in the USA is different to that in the EU. Unlike the EU, all aspects of device regulation fall to a single body—the Food and Drug Administration (FDA). Yet, in the USA, concerns have been expressed that the current system is suboptimal and leads to numerous device recalls and serious adverse events.8–10 Medical devices were responsible for 2712 deaths in the USA in 2006, double the number in 1997.11 A recent report of 113 recalled devices which caused serious health problems highlighted that most were approved using less stringent processes or were considered to be of such a low risk that they had been exempt from regulatory review.12 Approval of US devices takes more time, requiring more clinical data, and many companies now obtain approval in Europe first, often many years before the device appears on the US market.13
These rates of adverse medical device events and differences in regulatory approval suggest they are an important patient-safety issue worldwide, but there has been little evidence from the UK.14 Therefore, we aimed to describe the number of medical-device recalls in the UK and the clinical data requirements for regulatory approval, and determine the subsequent consequences of device recalls for patient safety.
Methods
For the purposes of this study, we defined a medical device using the European medical-device directives. These state: a medical device is any instrument, apparatus, appliance, software, material or other article, whether used alone or in combination, including the software intended by its manufacturer, to be used specifically for diagnostic and/or therapeutic purposes and necessary for its proper application. Additionally, the directives state that a medical device does not achieve its principal intended action in or on the human body by pharmacological, immunological or metabolic means, but it may be assisted by such means.2–4
We used information that is publicly available on the MHRA website (http://www.mhra.gov) to determine the number of devices which had been withdrawn or recalled over the 5-year period January 2006 to December 2010. Two forms of information were identified: field safety notices and medical-device alerts. Since 2006, the MHRA has also published field safety notices, which are issued by a manufacturer when a medical device needs to be recalled for technical or clinical reasons. In addition to this, the MHRA issues a medical-device alert as a means of communicating safety information to device end users in health and social care. Each medical-device alert is designated for ‘immediate action’ or ‘action,’ or provides updated information on previous alerts.
Because field safety notices and medical-device alerts do not state the CE class of the withdrawn devices, two authors (CH, MT) had to classify them independently into one of three CE-marked categories. This was done using the European Union directives for medical-device classification. Specifically, these are: Active Implantable Medical Device Directive, AIMDD (90/383/EEC),2 (General) Medical Device Directive, MDD (93/42/EEC)3 and the In Vitro Diagnostic Medical Device Directive, IVDMDD (98/79/EC).4 CE-device categories are as follows: Class I (generally regarded as low-risk devices); Class 2 (generally regarded as medium-risk devices); and Class 3 (generally regarded as high-risk devices). Where disagreement occurred in classification between the two authors, this was resolved by discussion.
We also categorised the agreed list of Class 3 devices by their main indication for use either by system (ie, cardiovascular, gastrointestinal, neurological or orthopaedic) or by mode (diagnostic, surgical instrument, in vitro device, infusion, imaging, radiotherapy, dialysis, sterilisation).
We also planned to determine the potential risk to patients associated with each of the withdrawn devices, using the system of classification employed by the FDA. The FDA system classifies risk of harm from a device using three levels:
a situation in which there is a reasonable probability that the use of, or exposure to, a product will cause serious adverse health consequences or death (FDA Class I);
a situation in which the use of, or exposure to, a product may cause temporary or medically reversible adverse health consequences or where the probability of serious adverse health consequences is remote (FDA Class II);
a situation in which the use of, or exposure to, a product is not likely to cause adverse health consequences (FDA Class III).
For medical-device alerts, we coded each device according to whether it was recalled or withdrawn from the market, and one author (MB) wrote to manufacturers of recalled devices to obtain further information. Specifically we asked for a copy of all field safety notices issued, the country where the CE marking was registered, the name of the notifying body, where the device was manufactured, where the device was packaged and details of clinical data that were submitted or in possession as part of the CE-marking process or data that have been published since the product was CE-marked (the questions are in online appendix 1). From this exercise, we learnt that manufacturers' clinical data were proprietary and therefore mainly not available for public scrutiny. Further to this, we contacted the MHRA by email to see if they held a central registry and/or clinical data for the CE-marked class 3 devices that had been recalled. They responded as follows:
The Medicines and Healthcare products Regulatory Agency (MHRA) does not have a definitive list of Class 3 medical devices, however, these are usually devices with the highest risk associated with their use, and are invasive, for example, heart valves, ICDs, implants, stents, etc.
Clinical data on these devices would be held by the manufacturer and is reviewed by the notified body before the product can be placed on the market. The MHRA does not routinely request or keep clinical data on medical devices.
We therefore contacted the six notifying bodies in the UK by email (online appendix 2) for information on Class 3 devices, who clarified:that all of the clinical data about medical devices they pass is unavailable to us. The notifying body is a client working on behalf of the manufacturer and sees the clinical data as being commercially sensitive.
Therefore, owing to insufficient data, we were unable to apply the FDA system of recalls to field safety notices. However, the two authors (CH, MT) were able to classify the MHRA Medical Device Alerts independently.12 Although the alerts are not exhaustive, they do contain a summary of the problem, the action to be taken and by whom, as well as the distribution list of the alert. Again, disagreements between the two authors were resolved by discussion.
We summarised data by year, by CE classification and by indication, and medical-device alerts by FDA recall system, presenting data as raw counts and proportions. Because the FDA recall system was undertaken only on a subset of the data, we calculated proportions and associated 95% CIs and used Cohen's kappa c as a measure of inter-rater reliability. We analysed data using Excel and SPSS version 17.
Results
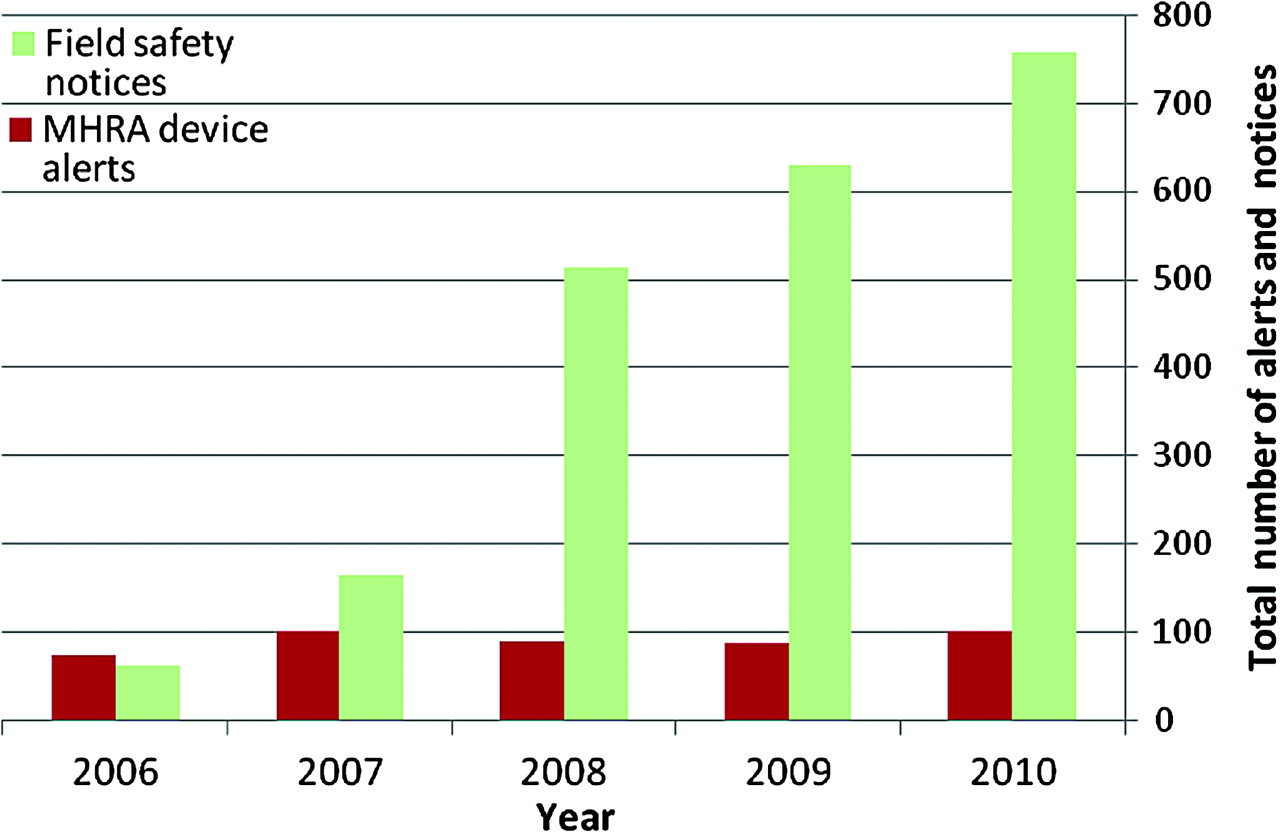
In total, there were 2124 field safety notices and 447 medical-device alerts issued in the 5-year period. While the numbers of medical-device alerts were consistent over this time period (range 73 to 100), the number of field safety notices increased by 1220% over the same period, from 62 in 2006 to 757 in 2010 (figure 1).

Total number of field safety notices and medical-device alerts per year (2006 to 2010).
Of the 2124 field safety notices, 327 (15.4%) were high-risk CE Class 3 devices, and more than half were related to the cardiovascular system (25%) or musculoskeletal system (33%). Table 1 shows there were 1527 (72%) medium-risk devices (CE Class 2) and 270 (12.7%) low-risk devices (CE Class 1).
Field safety notices by year and Conformité Européenne (CE) class of device
Of the 447 medical-device alerts, 147 (33%) devices were marked for immediate action, and 197 (44%) were to be withdrawn or recalled (table 2). We wrote to the manufacturers of 192 withdrawn devices whose contact details were listed on the alerts. We received 101 (53%) replies, of which only four (2.1%) provided the data requested: 21 replies provided partial answers, 11 declined to formally participate, 27 acknowledged the email but provided no response, and 38 emails bounced back because of an incorrect email address or an out-of-office reply.
Medical-device alerts by year, and Conformité Européenne (CE) class of device
Table 3 shows that 44% of medical-device alerts were assessed as having a reasonable probability of causing serious adverse health consequences or death, 38% caused temporary or medically reversible adverse health consequences, and 12.1% were assessed as not likely to cause adverse health consequences. The overall agreement between the two reviewers was moderate (Kohen kappa, 0.60);
Assessment of the relative degree of health hazard by Food and Drug Administration (FDA) classification of recalls for medical-device alerts
FDA classification of recalls
Recalls are classified into a numerical designation (I, II or III) by the Food and Drug Administration to indicate the relative degree of health hazard presented by the product being recalled.
Class I—a situation in which there is a reasonable probability that the use of, or exposure to, a product will cause serious adverse health consequences or death;
Class II—a situation in which the use of, or exposure to, a product may cause temporary or medically reversible adverse health consequences or where the probability of serious adverse health consequences is remote;
Class III—a situation in which the use of, or exposure to, a product is not likely to cause adverse health consequences.
Of the 447 medical-device alerts, 60 (13.4%) were classified as high-risk CE Class 3 devices, of which 53 (88%; 95% CI 80% to 97%) were assessed as having a reasonable probability of causing serious adverse health consequences or death (figure 2). Of the 53 devices, 16 (30%) alerts were notified as needing immediate action. For medium-risk devices, CE class 2b and 2a devices, 54% (44% to 64%) and 31% (23% to 38%) were judged as having a reasonable probability of causing serious adverse health consequences or death respectively. Figure 2 also shows that of the CE class 1 devices, those that carry the lowest risk, 34% (95% CI 26% to 42%) were assessed as a reasonable probability of causing serious adverse health consequences.

{kind=link}
{kind=link}
Food and Drug Administration (FDA) recall status of 447 medicines and Health Regulatory Authority (MHRA) device alerts by Conformité Européenne (CE) class of device.
Discussion
Main findings
We found a substantial increase in the number of field safety notices issued in the last 5 years by medical-device manufacturers, without concomitant increases in medical-device alerts issued by the MHRA. The number of field safety notices, which are issued by a manufacturer when a medical device needs to be recalled for technical or clinical reasons, increased by 1220% over the 5-year period, which represents a substantial concern for overall safety and impact on healthcare costs in the UK.
We were unable to access adequate clinical data or premarket approval data for recalled devices: only 2% of manufacturers were forthcoming in providing data. In the very few cases where we did receive data, these were mainly literature reviews and were not comparable with systematic reviews.
In addition, we found that nearly half of medical-device alerts were related to devices that had a reasonable probability of causing serious adverse health consequences or death. Moreover, of the most risky devices (CE Class 3 devices) that were recalled, nearly nine out of every 10 were judged independently by two clinicians to have a reasonable probability of causing serious adverse health consequences or death based on the information contained in the MHRA medical-device alerts. However, it was not unusual for us to find low-risk devices, CE class I devices, leading to potentially serious adverse events; numerous defective wheelchairs and hoists needed to be recalled, which potentially has led to considerable morbidity.
Implications
A substantial number of important issues arise because of our findings. First, why are field safety notices rising dramatically, while medical-device alerts are not? In 1999, the UK Medical Device Agency published only eight advice notices, eight device alerts and 36 safety notices.7 One reason is that medical device numbers have increased substantially over time. One could also argue that manufacturers are doing their job; the question then is: how many device alerts could or should there be? Collation of medical-device alerts associated with a reasonable probability of causing serious health problems or death from US FDA, UK MHRA, Health Canada and manufacturers resulted in a total of 1588 alerts in 2010 alone.15
Second, the current system of CE marking is confusing. For instance, while contact lens cleaners are CE Class 2b devices, contact lenses could be classified as Class I. Manufacturers ultimately decide on the class of the device and therefore the level of clinical data required. We found the difference between Class 2a and 2b often difficult to determine or justify on clinical grounds alone. A new directive (2007/47/EC) that came into effect in March 2010 highlights this confusion. For instance, it states: ‘the central circulatory system now includes the vessels aortic arch and descending aorta to the aortic bifurcation,’ whereas in the previous directives it did not. Devices in contact with these vessels will now be considered high-risk, whereas, one can only surmise, in the past they were deemed at a lower risk and subject to less stringent data requirements at the regulatory stage.
Third, are the requirements for preapproval clinical data fit for purpose in Europe? Preapproval data for medical devices in Europe do not require demonstration of efficacy. Clinical data used for CE marking may be either a review of the relevant scientific literature or the results of a clinical study. The first of these is used by the majority of manufacturers of low- to medium-risk devices (Class I, 2a and 2b). Where clinical data are used, they may be unpublished, or data generated on an equivalent device.16 Unlike pharmaceutical regulation, no summaries are publicly available for independent assessment.
It is particularly concerning that we were unable to review any of the clinical data provided to achieve a CE mark, as the relevant data are held by the company or the notifying bodies and not a public body, such as the MHRA. This means that they are not subject to the Freedom of Information Act, a means by which researchers can access information. Again, this contrasts markedly with the situation for drugs, where this information can be obtained: the European Medicines Agency policy has agreed to give access to all business-related documents on medicines for humans. Surveillance of devices in practice is therefore lax,17 and whereas we can access mortality data in adult cardiac surgery for named surgeons,18 we cannot currently do the same for named devices in many different specialities and systems.
Finally, how should the current system of regulation be changed? Since device recalls will continue to occur and seem to be increasing, better communication of the risk of the recall should be considered, to help clinicians and patients make decisions that allow appropriate risk assessment.8 Among the new directives' (2007/47/EC1) requirements is the need for more clinical data and more frequent clinical investigations; these data should be made publicly available, particularly for the highest-risk devices, so that end users have clear evidence on which to base important (and potentially costly) decisions regarding device replacement or recall. In addition, adoption of the US system of recall class, at the time of the device alert would allow a better understanding of the potential magnitude of the safety problem. Finally, because the highest-risk devices come from cardiovascular and musculoskeletal systems, these areas should be prioritised for independent national registries of implantable devices with publicly accountable data.
Limitations
The main limitation of the study was the lack of available data on details of device withdrawals, or quantification of the number or current use of devices affected. Our CE classification of devices and FDA recall status was therefore based predominantly on our clinical experience based on the information that was publicly available, and this judgement may therefore differ from manufacturers' classification and from other clinicians. Owing to a lack of data made available to us, we were also unable to determine the reason for the rise in field safety notices and therefore cannot speculate on when the problem first arose and, more importantly, what kind of clinical testing had been undertaken prior to the device going on the market. The absence of a central registry containing information on how many devices are currently in use in the UK limited our ability to fully assess the implications of our findings on patient safety. This means that we are unable to quantify the true number of patient harms caused by medical device recalls. Finally, we are unable to determine which of the safety alerts were acted on by the healthcare and social care community, the proportion of patients (and/or devices) who needed to be traced, and the workload and costs involved in these actions.
Conclusions
The size and scale of the medical device recalls substantially impact on NHS workload and patient safety, and the number of field safety notices continues to grow. Significant problems exist in the UK with a lack of access to transparent data and a registry of the highest-risk devices, which prevents a full understanding of the size and impact on patient safety.
Footnotes
To cite: Heneghan C, Thompson M, Billingsley M, et al. Medical-device recalls in the UK and the device-regulation process: retrospective review of safety notices and alerts. BMJ Open 2011;1:e000155. doi:10.1136/bmjopen-2011-000155
Funding CH and MT have received payment for evidence-based training workshops.
Competing interests DC and MB are employees of the BMJ Group. MT is an editorial adviser who attends the BMJ's research manuscript committee from time to time, for which he receives an honorarium and, when in the UK and attending in person, travel expenses. CH and MT co-developed products with the BMJ Evidence Centre and run a joint conference with the BMJ Evidence 2011.
Contributors CH and DC devised the study. CH and MT extracted the data for the Field Safety Notices and Medical Device Alerts. MB wrote to manufacturers, Notifying Bodies and the Medicines and Health Regulatory Authority. CH and MT analysed the data, and all authors contributed to the writing of the paper and approved the final draft.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement All data and files used in the manuscript are available upon request from the corresponding author.