Article Text

Abstract
Objectives This review investigates characteristics of implemented adaptive design clinical trials and provides examples of regulatory experience with such trials.
Design Review of adaptive design clinical trials in EMBASE, PubMed, Cochrane Registry of Controlled Clinical Trials, Web of Science and ClinicalTrials.gov. Phase I and seamless Phase I/II trials were excluded. Variables extracted from trials included basic study characteristics, adaptive design features, size and use of independent data monitoring committees (DMCs) and blinded interim analyses. We also examined use of the adaptive trials in new drug submissions to the Food and Drug Administration (FDA) and European Medicines Agency (EMA) and recorded regulators’ experiences with adaptive designs.
Results 142 studies met inclusion criteria. There has been a recent growth in publicly reported use of adaptive designs among researchers around the world. The most frequently appearing types of adaptations were seamless Phase II/III (57%), group sequential (21%), biomarker adaptive (20%), and adaptive dose-finding designs (16%). About one-third (32%) of trials reported an independent DMC, while 6% reported blinded interim analysis. We found that 9% of adaptive trials were used for FDA product approval consideration, and 12% were used for EMA product approval consideration. International regulators had mixed experiences with adaptive trials. Many product applications with adaptive trials had extensive correspondence between drug sponsors and regulators regarding the adaptive designs, in some cases with regulators requiring revisions or alterations to research designs.
Conclusions Wider use of adaptive designs will necessitate new drug application sponsors to engage with regulatory scientists during planning and conduct of the trials. Investigators need to more consistently report protections intended to preserve confidentiality and minimise potential operational bias during interim analysis.
- adaptive design
- flexible design
- clinical trial
- review
- history
- regulation
- policy
- FDA
- EMA
- data monitoring committee
- interim analysis
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
- adaptive design
- flexible design
- clinical trial
- review
- history
- regulation
- policy
- FDA
- EMA
- data monitoring committee
- interim analysis
Strengths and limitations of this study
This review explores the use of adaptive designs in published and publicly available trials in a broad set of databases, although our search terms do not capture adaptive trials not described as adaptive by trial sponsors.
We have affirmed the findings of existing reviews and have examined some adaptive trial features not previously described.
We have examined the use of adaptive design trials in submissions to both the Food and Drug Administration and European Medicines Agency.
Our lack of access to non-public regulatory review materials prevented us from locating more adaptive trials, or additional details of the trials, used in regulatory evaluation of new products.
Introduction
Well-conducted randomised controlled trials (RCTs) have long been recognised as the gold standard for assessing efficacy of clinical interventions, valued for their statistical rigour and mechanisms to avoid bias. Regulatory agencies around the world generally prefer RCTs when evaluating whether to authorise marketing of new drugs. Yet, modern RCTs can demand substantial time and resources.1
Adaptive designs have been developed as an alternative to traditional RCT design. Traditional RCTs tend to allocate patients to control and intervention groups according to a consistent randomisation scheme throughout a trial. In adaptive trials, patient outcomes are observed and analysed at predefined interim points and predetermined modifications to study design can be implemented based on these observations. In late 2016, the US Congress passed into law the 21st Century Cures Act, which instructs the Food and Drug Administration (FDA) to update its guidance on adaptive designs for sponsors of investigational drugs and biological products. The legislation refers to adaptive designs as ‘modern’ and ‘novel’ methods.2 Some adaptive methods are indeed recent developments, while others have existed for decades and have had a complex history. There was a growth in biostatistics literature on adaptive designs in the 1960s and 1970s, but initially adaptive methods were, as one observer described, ‘almost totally unused in practice’.3 4 As scientists and regulators gradually gained experience with adaptive trials, they encountered some challenges with implementing and interpreting some types of adaptations.5–9 Common recurring critiques have included increased risks of falsely detecting treatment effects, prematurely discarding promising therapies and causing statistical or operational bias.5 10–12 Still, interest in adaptive designs grew, with new methods continuing to be explored. Some segments of the scientific community, as well as the pharmaceutical industry, have promoted adaptive designs to potentially make Phase II and Phase III registration trials more efficient, informative, or more likely to demonstrate a drug effect.13–16
Recent studies have identified challenges that have limited the use of adaptive designs, such as lack of applied training and inadequate information regarding completed adaptive design trials, leading to deficiencies in the practical understanding of adaptive trial implementation.17 18 Other studies have provided important insights into the characteristics of adaptive design trials to date.19–21 We endeavoured to complement and expand on the findings of these studies to provide additional information for continuing policy development. To do so, we conducted a review of publicly available adaptive trials and their characteristics. We compared our results with trends in standard RCTs where possible to investigate variations between adaptive and standard trials. Finally, we examined the use of adaptive trials identified in this review in the drug approval process in the USA and EU.
Methods
Data sources and searches
We conducted systematic searches of EMBASE, PubMed, Cochrane Registry of Controlled Clinical Trials and Web of Science databases in September 2014 using phrases in English derived from descriptions of the 10 most common forms of adaptive designs: adaptive hypothesis, adaptive treatment-switching, biomarker adaptive, adaptive dose-finding, pick-the-winner/drop-the-loser, sample size re-estimation, adaptive randomisation, adaptive group sequential, adaptive seamless and multiple adaptive.22 23 Table 1 includes definitions for each of these types of adaptive designs. We also included adaptive designs that did not seem to fit any of these specific categories, but that fit the FDA’s definition of adaptive designs as prospectively planned modifications to study design or hypotheses based on analysis of interim data from subjects in the study.5 We did not limit the searches to any specific date ranges and included all available adaptive trials for our study at the time of our research.
Definitions of types of adaptive designs23
We enriched these results with a second, iterative search in the same databases (excluding Web of Science) in October 2014 using phrases extracted from the first search results. This second search was designed to capture additional studies using different and broader search terms.
We did not include Web of Science in the supplemental review, as Web of Science is an automated search programme that captures results more loosely across all scientific disciplines. The second set of broader search terms captured an excessive number of irrelevant Web of Science results beyond our resources for review and so were not included in this analysis. We also manually mined references of selected articles to identify missed trials (none were found).
Finally, to enlarge the sample size of our study to better capture and analyse variables of interest, we searched ClinicalTrials.gov in June 2015 using a similar search architecture. See online supplementary appendix, pages 1–3 for specific search phrases, strategy details and definitions of adaptive design types. It is beyond the scope of this paper to review unpublished adaptive trials not included in ClinicalTrials.gov or adaptive trials that fall outside of the definitions provided by Chow and Liu and the FDA.5 22 23
Supplementary file 1
Study selection
We excluded studies not involving human subjects, systematic reviews, meta-analyses, conference abstracts, commentaries, editorials or statistical methods or economics discussions. We excluded incomplete trials in progress to avoid misrepresenting trials, which can sometimes change format while underway. Several variables explored in this study are also only determinable for completed trials and so excluding trials in progress enabled us to keep our sample roughly consistent. We excluded Phase I and seamless Phase I/II trials because such exploratory trials usually have low impact on regulatory approval and/or clinical uptake of treatments.5 See online supplementary appendix, pages 3–4 for a more detailed description of trial inclusion/exclusion rationales.
Two authors (LEB and ASK) separately reviewed 10% of results and compared assessments. The Cohen kappa coefficient assessment of inter-rater agreement was 0.91. One author (LEB) applied the inclusion criteria to remaining trials.
Data extraction and quality assessment
Two reviewers (LEB and NFK) extracted data and checked each other’s work for accuracy. Disagreements were resolved by a third reviewer (ASK).
Data synthesis and analysis
We recorded basic study characteristics including the year of trial publication (or completion on Clinicaltrials.gov), adaptive design type, research phase (II, seamless II/III, III, IV), trial duration, subject sample size and patient demographics (age, race, ethnicity, gender).
We also examined the nature of research in each adaptive trial, including type of disease studied in a trial; whether the disease was a rare condition according to the National Institutes of Health Office of Rare Diseases Research classification system or FDA orphan drug designation system; whether mortality was an endpoint; types of trial endpoints (surrogate, clinical or both); intervention type (drug, medical device or other) and whether the experimental intervention was found effective or ineffective.
We included trial location and author location, noting adaptive trials conducted outside North America or Western Europe while having authors from these locations (a marker of trial outsourcing). Author affiliations and funding sources were recorded.
We noted whether articles mentioned independent data monitoring committees (DMCs) or blinded interim analyses. We also classified whether adaptive methods were ‘well-understood’ or ‘less well-understood,’ based on definitions in the FDA Guidance for Industry on adaptive designs. The FDA defined ‘well-understood’ as designs employing relatively well-established methods with limited risk of introducing bias or impairing study interpretability and ‘less well-understood’ as designs with which there is relatively little regulatory experience and that may risk problems such as falsely detecting treatment effects, introducing bias, or causing inconsistency between trial hypotheses and statistical tests.5 Trials that used only the seamless Phase II/III design were not evaluated as well-understood or less well-understood, because the FDA did not include the Phase II/III design in its categorisation of trials as well-understood or less well-understood.
We assessed how adaptive trials have been used by regulatory agencies. Because the vast majority of trial authors were located in the USA or Europe and because the FDA and European Medicines Agency (EMA) are leading global drug regulators, we examined whether each trial was in online product review packages of these agencies.24 25 For trials that were in online review packages, we recorded any regulatory reviewers’ commentary on the adaptive methods. We then determined whether adaptive trials in review packages were final or pivotal trials used for product evaluation. We recorded the length of time required by the FDA and EMA to review applications, whether the products were approved for the purposes studied in the adaptive trials and whether they received FDA or EMA orphan designation (incentive programmes to encourage research on rare diseases for which sponsors otherwise may not recover research costs.) Finally, we recorded whether products received FDA priority review or EMA accelerated assessment (expedited review systems for drugs of major interest to public health). See online supplementary appendix, pages 4–6 for definitions of extracted variables.
For variables unavailable or inconsistently reported on ClinicalTrials.gov (see online supplementary appendix, pages 6–7), we only evaluated literature review results. We also compared our findings with trends in standard clinical trial publication volume,19 study location,26 27 participant demographics,28 funding,27 29 use of blinded interim analyses and independent DMCs28 and regulatory review time.30 31 High-quality data were unavailable on trends in intervention types, participant race or ethnicity, average sample size or duration of standard trials. So, we conducted a systematic sampling of published standard RCTs for general comparisons. One author (LEB) selected 50 standard RCTs per 5-year period (approximately) from 1976 to 2014, recording trends in types of interventions and subject race, which were then qualitatively compared with types of interventions and subject race in adaptive trials. To compare sample size and duration between the standard RCTs and adaptive trials by phase, subsets of 50 Phase II and 50 Phase III standard RCTs were selected from the systematic sample. These sets of standard RCTs were selected from the years in which adaptive trials appeared, sampling the same number of standard trials by phase per year as adaptive trials by phase appeared in our literature review. For each year sampled, the first available Phase II and Phase III standard RCTs were taken. Further details on the methods of our supplementary sampling of standard RCTs are described in the online supplementary appendix, page 7.
Results
Our protocol retrieved 2711 results, from which we identified 142 trials for analysis (figure 1). Ninety-nine were published articles and 43 were independent ClinicalTrials.gov listings.

Flow diagram derived from preferred reporting items for systematic reviews and meta-analyses (PRISMA). This diagram reports results of a search for published adaptive design clinical trials in Web of Science database on 17 September 2014; in PubMed, EMBASE and Cochrane Registry of Controlled Clinical Trials databases on 22 October 2014 and in ClinicalTrials.gov on 20 June 2015.
Types of adaptive trials
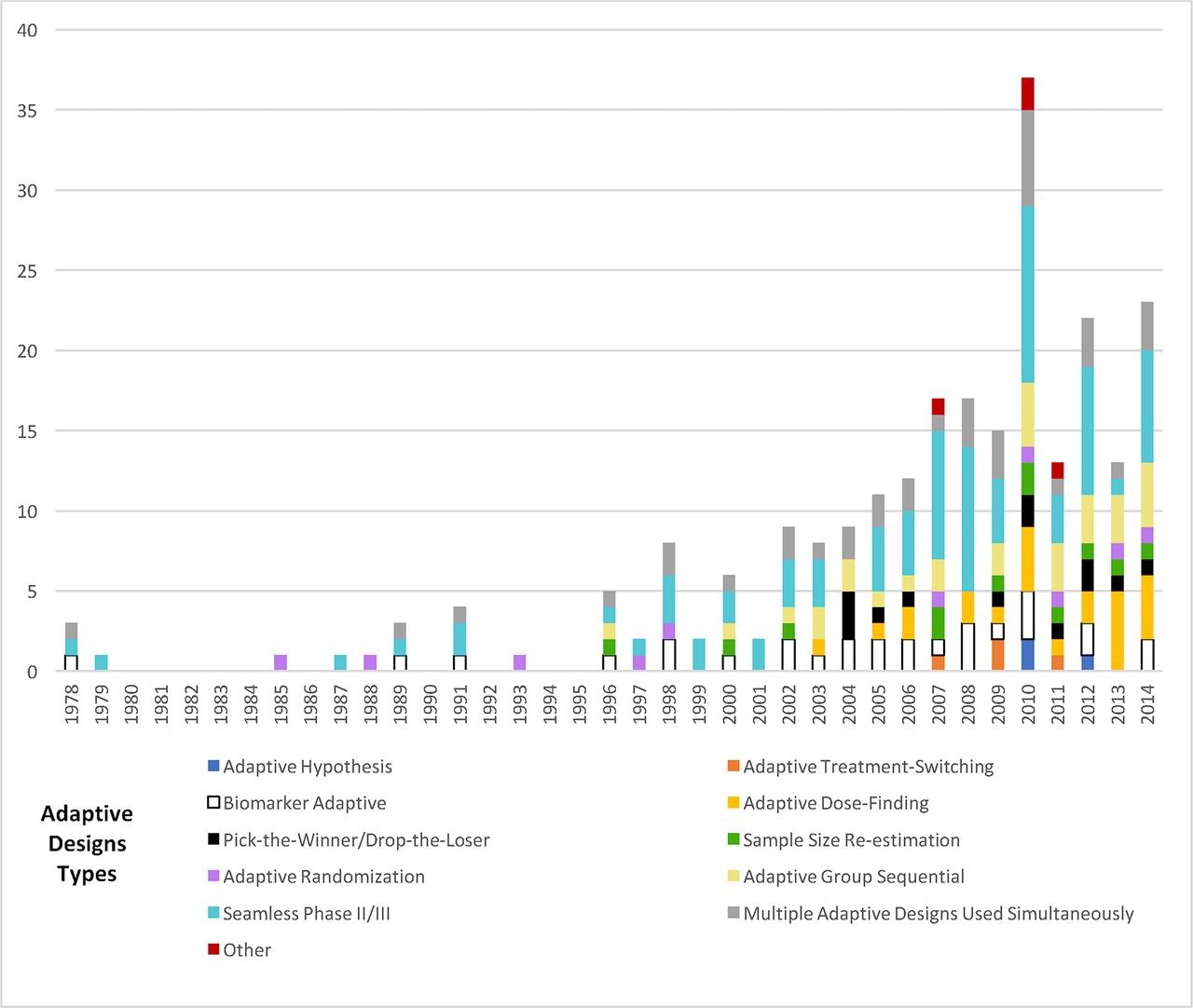
Adaptive trials expanded in the scientific literature since the mid-1990s. The most frequently appearing type of adaptation was the seamless Phase II/III design 81/142 (57%), followed by adaptive group sequential 30/142 (21%), biomarker adaptive 28/142 (20%), adaptive dose-finding 23/142 (16%), pick-the-winner/drop-the-loser 13/142 (9%), sample size re-estimation 11/142 (8%), adaptive randomisation 10/142 (7%), adaptive treatment-switching 4/142 (3%) and adaptive hypothesis 3/142 (2%). Numerous trials incorporated multiple adaptations (figure 2).

Prevalence of adaptive design type in surveyed trials. Adaptive trials first appeared in ClinicalTrials.gov search results in 2002; data prior to 2002 reflect only literature review results and data after 2002 reflect combined literature review and ClinicalTrials.gov results.
Approximately one-quarter of all trials were Phase II 33/142 (23%), while 13/142 (9%) were Phase III and 5/142 (4%) were Phase IV. Trial phase was unclear in 10/142 (7%) of studies. A variety of adaptive designs appeared in all phases of research, although adaptive randomisation only occurred in Phase II trials.
Trial design characteristics
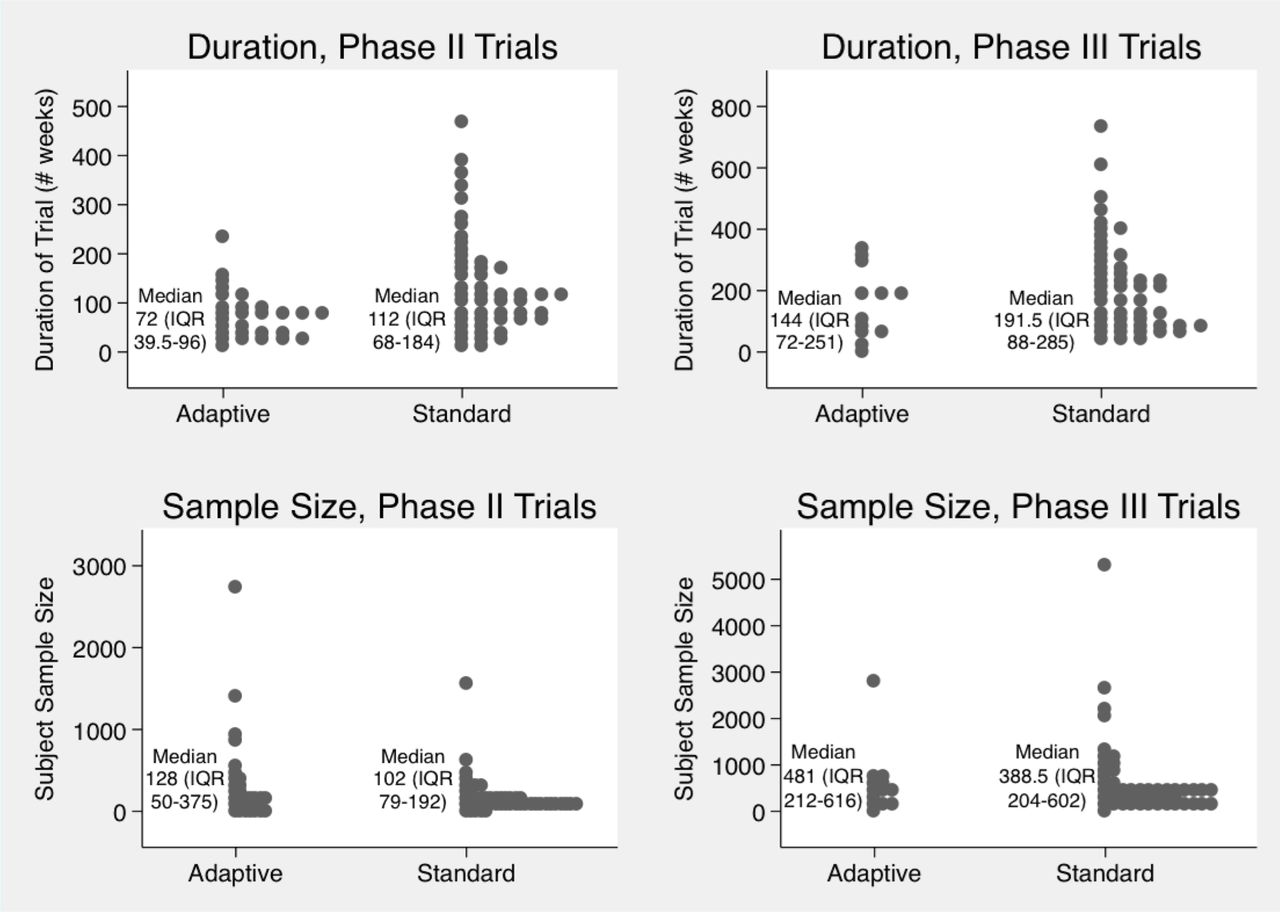
Data were available on duration for all but four Phase II trials and one Phase III trial. Figure 3 depicts the duration and number of participants in our samples of adaptive trials and standard trials.

Duration and sample size of adaptive and standard Phase II and III trials. Phase II and III adaptive and standard trials, including median durations in weeks, as well as median participant sample sizes. Each dot represents a trial.
To further compare duration and sample size among subsets of similar adaptive and traditional trials, we matched sets of adaptive trials with traditional RCTs of the same phases testing similar therapeutics for the same conditions, finding no substantial differences in duration or sample size between the traditional and adaptive trials (see online supplementary appendix, pages 7–8 and online supplementary appendix table 1).
Patient demographic data were inconsistently reported in adaptive trials. Most trials 132/142 (93%) included data on participants’ age, while 53/142 (37%) reported participant race and/or ethnicity. Table 2 depicts the number of adaptive trials including participants of different age groups and, out of the trials that reported participant race/ethnicity, the number of trials that included participants of different racial/ethnic demographic groups.
Trials reporting participant age and race/ethnicity
Five trials included only men and 15 trials included only women. In trials with both genders, men were more represented, as 61 trials included more men than women, 51 included more women than men and 17 trials included approximately equal numbers of men and women. Adaptive trial demographic trends were consistent with those of standard clinical trials.
Table 3 illustrates disease areas most frequently examined and endpoints measured in the adaptive trials. One-quarter of adaptive trials investigated rare diseases. Mortality was an endpoint in 62/142 (44%) of trials. Surrogate measures were frequently used as outcomes in adaptive trials, alone or in combination with clinical endpoints. However, adaptation decisions were based on surrogate measures in only 28/142 (20%) of the trials.
Leading areas of investigation, endpoints and intervention types in adaptive trials
Many adaptive trials (121/142; 85%), tested drugs; 13/142 (9%) tested other therapies such as vaccines, diet or treatment delivery methods; 9/142 (6%) tested medical devices. One trial tested a treatment qualifying as both a drug and an ‘other therapy’ (table 3). Roughly similar to adaptive trials, most trials in our systematic sampling of standard RCTs (85%) tested drugs, 14% tested other interventions and 1% tested medical devices. In 49/99 (49%) of published adaptive trials, the experimental interventions were deemed efficacious.
Location and sources of adaptive trials
Approximately one-quarter of adaptive trials 40/142 (28%) only enrolled patients in Western Europe, one-quarter (34/142; 24%), only enrolled patients in the USA and 28/142 (20%) enrolled patients at multinational trial sites. This patient location diversity approximates that of traditional RCTs. Trial authors were often located in the nations in which trials occurred. However, for 29/99 (29%) of published adaptive trials, authors were from the USA or Western Europe while trial sites were outside these locations. This was similar to traditional RCT trends of investigators offshoring trials or engaging in collaborative multinational trials.
Published adaptive trials often had multiple authors with diverse institutional affiliations. Nearly all trials had at least one author affiliated with an academic medical centre or hospital (97/99; 98%). Half had at least one author affiliated with industry (50/99; 50%), 37/99 (37%) had at least one author affiliated with a private foundation and 2/99 (2%) had at least one author affiliated with government. Most first authors listed affiliation with an academic medical centre or hospital (91/99; 92%) and 8/99 (8%) listed affiliation with industry.
Two-thirds (92/142; 65%) of trials reported industry funding, 27/142 (19%) reported government funding, 14/142 (10%) private foundation funding, 10/142 (7%) academic medical centre funding and 20/142 (14%) did not list funding source (figure 4). Trials sometimes listed funding from multiple sectors, while the proportion of trials reporting industry funding grew over time. Funding patterns for adaptive trials were consistent with general clinical research funding patterns.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Sources of funding for adaptive design clinical trials. Number of trials receiving funding from each type of source. Data prior to 2002 reflect only literature review results and data after 2002 reflect combined literature review and ClinicalTrials.gov results.
Data monitoring, interim analysis and FDA classifications of adaptive methods
In published adaptive trials, about one-third (32/99; 32%) mentioned an independent DMC. Six out of 99 (6%) mentioned blinded interim analysis. Blinding methods varied; in 3/99 (3%) of trials, authors simply reported blinded interim analysis without further specification. In another 3/99 (3%) of trials, investigators, sponsors and patients were blinded from results of interim analyses, and DMCs reviewed partially blinded interim analyses. For example, in one study an independent statistician calculated interim results, sharing only broad results with the DMC, identifying specific treatment groups to the DMC by letters A to G rather than actual treatments.32
Reporting rates of independent DMCs and blinded interim analyses were on par with traditional RCTs; in one study of 291 traditional RCTs published in top tier journals, 35% reported independent DMCs and 2% mentioned blinded interim analysis.28
A minority 7/57 (12%) of evaluable published trials used adaptive designs that qualified as ‘well-understood’ by the FDA. Well-understood and ‘less well-understood’ adaptive designs were used across Phase II and Phase III trials. The single published adaptive Phase IV trial used a less well-understood design.
Use of adaptive trials by US and EU regulators
We found that 13/142 (9%) of adaptive trials in our sample were used for FDA product approval consideration, and 17/142 (12%) were used for EMA product approval consideration. A slightly smaller fraction, 11/142 (8%), of adaptive trials were the final or pivotal trials used for FDA approval consideration; 7/142 (5%) were final or pivotal trials used for EMA approval consideration. The remaining trials evaluated drugs that had already been approved, treatments not requiring approval or treatments for which approval packages were not available online.
The FDA spent a median of 12.2 months reviewing new drug applications that included adaptive trials and the EMA spent a median of 14 months for such reviews. Review times were roughly similar to some published estimates of standard FDA review times for the years covered and slightly longer than other estimates of FDA and EMA review times by 6–7 weeks. During the review process, the FDA and EMA used adaptive trials to evaluate new products in some cases without reporting major problems. However, in other cases, the agencies listed some specific concerns regarding the adaptive designs deployed, including lack of sufficient statistical power to properly evaluate the experimental treatments, risk of ineffectively evaluating doses, risk of falsely detecting treatment effects, inadequate blinding and inappropriate DMC activity. In some applications to both the FDA and EMA, regulators found the sample sizes too small in adaptive trials to effectively interpret the results or draw robust conclusions. In one example, after extensive review, the EMA found an adaptive group sequential trial testing an herbal treatment for acute rhinosinusitis to provide insufficient data. The adaptive design involved early stopping with reduced sample size, claiming to demonstrate treatment efficacy, but the EMA found the reduced sample size problematic and contended that more trials were needed before conclusions could be drawn regarding the treatment.33 Several separate regulatory reviews stated that adaptive trials with smaller sample sizes failed to gather sufficient data on subpopulation effects, such as outcomes by race or gender. Many applications with adaptive trials had extensive correspondence between drug sponsors and regulators regarding the adaptive designs, in some cases with the regulators requiring revisions or alterations to research designs prior to product approval. In several cases in which trial sponsors had limited advanced communication with regulators, there were lengthy post-trial communications over concerns in adaptive trial review.
In our sample, 13/142 (9%) of adaptive trials tested drugs that were found to have been approved by the FDA through reliance in part on the adaptive trial, while 10/142 (7%) of adaptive trials tested drugs that the EMA approved using the adaptive trial. Nine out of 142 (6%) adaptive trials studied treatments that were found to have received FDA orphan drug status while 6/142 (4%) studied treatments that received EMA orphan drug designation. Three out of 142 (2%) adaptive trials were included in new drug applications that received FDA priority review; none received EMA accelerated assessment. See online supplementary appendix table 2 for information on published adaptive trials that were used for EMA and FDA approval consideration.
Discussion
In this review, while we found that many adaptive methods have existed for decades, there has been a recent growth in publicly reported use of adaptive designs among researchers around the world, particularly among industry-funded researchers and investigators located in North America and Europe. Adaptive trials have been used to study a range of diseases and interventions (mostly drug treatments), overlapping with the traditional roles of RCTs in many ways. Adaptive designs have been deployed most prominently in oncology and have relied on a combination of surrogate measures and clinical endpoints. A minority of adaptive trials reported blinded interim analyses and use of independent data monitoring committees. The FDA and EMA have had mixed experiences with adaptive designs.
Adaptive design methodology has been the subject of several recent reviews. Extant work has explored general features of adaptive trials collected from a variety of timeframes,17 34 regulatory institutions including the Center for Biologics Evaluation and Research (CBER) and the Center for Devices and Radiological Health (CDRH) at FDA,20 21 and broad databases including ClinicalTrials.gov.19 In addition, various studies have explored specific aspects of adaptive trials, including perceptions of adaptive designs and barriers to their implementation,17 18 35 36 the explicit or implicit reporting of adaptive designs in oncology,37 and whether adaptive methods have been prospectively planned or designed ad-hoc.37 Our study augments these existing works and our results largely affirm their findings. We also investigated several other policy-relevant variables related to the design of publicly available adaptive trials, such as trial participant demographics, explicit reporting of blinded interim analyses, and independent DMCs.
FDA and EMA regulatory experience with adaptive designs remains a topic of widespread interest.20 35 38 39 Our results support the findings identified in other surveys in this area, most notably by identifying recurring regulatory concerns over trial interpretability.20 In other publications, FDA and EMA investigators reported more adaptive trials than we found in our database searches, indicating that adaptive designs may be more widespread in regulatory submissions than the number of trials that can be publicly located using a range of pertinent search terms.20 21
In our review, we encountered numerous cases in which regulatory officials found methodological deficiencies or data collection problems specific to the adaptive designs, resulting in lengthy correspondence with trial sponsors and complications in the review process. Therefore, it may be useful for policies related to adaptive designs to encourage new drug application sponsors to engage closely with regulatory scientists before and during trial conduct, to reduce the likelihood of repeating past scenarios in which regulators found results from adaptive trials insufficient.18 21 35 In particular, trials including multiple adaptations have not been well-understood and so investigators may wish to exercise some caution with multiple adaptations in a single trial, although this may change in the future.
Numerous adaptive trials, like standard randomised controlled trials, have been international collaborations, which has advanced the collection of data on geographically diverse patients before products are approved by major global regulators. Adaptive trials have also recruited patients with race, ethnicity, age and gender representation approximately in the same proportions as standard trials. However, it is concerning that in some cases, regulators found that reduced patient sample sizes in adaptive trials resulted in inadequate numbers of diverse participants. To encourage representation of a wide range of populations in adaptive trials, future revised regulatory guidelines should specify that adaptive designs should not reduce trial sample sizes in ways that prevent evaluation of treatment outcomes among these populations beyond what could be reasonably expected of standard trial designs for the same investigations.
Finally, we found low reporting of blinded interim analyses and independent data monitoring committees in adaptive trials. Reporting rates were commensurate with such rates for traditional trials; in 1 study of 291 traditional RCTs published in top tier journals, 35% reported independent DMCs and 2% mentioned blinded interim analyses.28 However, it has been suggested that blinded interim analyses and independent data monitoring committees are particularly important for adaptive trials because DMCs advise on the implementation of adaptations during interim analyses in adaptive trials. Statisticians and regulatory experts have strongly recommended establishing safeguards to maintain interim analysis blinding from investigators, sponsors or anyone outside DMCs to avoid introducing bias into ensuing study design, conduct, or interpretation.5 39 40 Scientists in academia, industry, the FDA, and the EMA have also noted that adaptive trials without independent DMCs are at greater risk of biased data interpretation.5 14 41 42 Trials in this review may have incorporated blinded interim analyses and independent DMCs without stating so, although specification of these features would be beneficial when possible, given the general scientific consensus on their importance.5 42 Therefore, it may be useful for policy makers and investigators to promote increased reporting of independent DMCs and blinded interim analyses when possible and appropriate in adaptive trials. Given the reasonable concerns regarding operational bias, investigators may also wish to exercise caution when using adaptive designs in scenarios for which blinded interim analyses or independent DMCs do not seem feasible. A promising recent proposal of a CONSORT extension for adaptive designs includes the recommendation that adaptive trials include detailed descriptions of mechanisms to minimise/control for operational bias in interim analysis.36 The use of this CONSORT extension could be helpful in stimulating improved reporting of independent DMCs and blinded interim analyses. Additionally, given that many published trials used adaptive designs that qualified as ‘less well-understood’ at the time of publication, improved reporting guidelines may help eliminate areas of confusion for regulators and reviewers of adaptive trials and could potentially be useful toward more adaptive design trials being considered ‘well-understood.’
There were several limitations to our study. Our searches and manual review did not retrieve all adaptive trials. As there is some ambiguity regarding what qualifies as adaptive, we may have missed trials not meeting the definitions described above or uncommon or contested views of adaptive designs.43 44 We did not capture trials that were not completed or unpublished trials unavailable in ClinicalTrials.gov, and our search only captured trials self-identified as adaptive. While we limited our inclusion criteria for adaptive group sequential trials to include only group sequential trials that would have been universally considered by their authors and all scholars at the time of their publication to be adaptive by allowing for adaptations beyond stopping or continuing at interim analysis (see supplementary appendix, pages 3–4), we understand that many scholars and regulators now view all group sequential trials as adaptive. From this latter perspective, our study would have revealed a smaller number of group sequential trials. It cannot be assumed that our findings describe trends in all adaptive trials, but rather trends in self-identified adaptive trials that investigators have publicly registered and/or deemed appropriate for publication. As a supplemental analysis, we reviewed frequency of adaptive trials as a percentage of published clinical trials overall since the 1970s and as a portion of trials submitted to leading medical journals, finding adaptive trials relatively uncommon using these methods (see supplementary appendix, page 10). Other search approaches can focus on particular types of adaptive trials.45 Mistry et al 37 reviewed RCTs in oncology in a single year to locate designs that were referred to as adaptive, as well as adaptive trials that were not self-identified. They concluded that adaptive design use should be better reported, affirming similar suggestions by other scholars. These studies align with our contention that it is currently difficult to capture all published adaptive trials. Our exploration of regulatory reviews of adaptive trials is limited to publicly available agency documents on self-identified adaptive trials and may not capture all examples of adaptive trials used for regulatory purposes. Additionally, our comparisons of duration and sample size between adaptive and traditional trials only provide a snapshot of some recent experiences. Assessing the meaning of differences in trial duration and sample size between adaptive and traditional trials requires the studies to be designed in exactly the same way, varying only by using adaptive or traditional designs. Opportunities for such comparisons are rare, although they have been modelled.45
Despite these limitations, our results provide a broad review of trends in adaptive design trial characteristics, investigated endpoints, and their reception by regulatory agencies in the USA and Europe. As adaptive designs are not widely deployed, regulators and scientists are still exploring how and the extent to which they may be incorporated into the evaluation of experimental therapies. Future investigations of adaptive designs could examine ongoing dynamics in trials as new regulatory guidelines are established.
Acknowledgments
The authors thank Danielle Piacentile (Dartmouth College) for assistance with data collection for confirmatory analyses in the Appendix, Sara Dejene (Division of Pharmacoepidemiology) for ClinicalTrials.gov data collection assistance and Julie Whelan, MS (Countway Library) for database search assistance.
References
Footnotes
Contributors LEB, ASK: wrote/revised manuscript. LEB, ASK, JA: designed research. LEB, NFK: performed research and analysed data.
Funding This work was supported by the Harvard Program in Therapeutic Science and a Brigham and Women’s Hospital Innovation Hub grant (no grant numbers). Dr Kesselheim’s work is also supported by a grant from the Laura and John Arnold Foundation with additional support from the Engelberg Foundation.
Competing interests All authors have completed the ICMJE uniform disclosure form at www.icmje.org/coi_disclosure.pdf and declare: ASK had financial support from the Laura and John Arnold Foundation and Harvard Program in Therapeutic Science and LEB had support from Brigham and Women’s Hospital Innovation Hub for the submitted work; no financial relationships with any organisations that might have an interest in the submitted work in the previous three years; no other relationships or activities that could appear to have influenced the submitted work.
Patient consent Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement This review draws on publicly available trials and regulatory review documents only. No new data were generated in undertaking this study.