Article Text

Abstract
Introduction Management of thumb base osteoarthritis (OA) using a combination of therapies is common in clinical practice; however, evidence for the efficacy of this approach is lacking. The aim of this study is to determine the effect of a combination of conservative therapies for the treatment of thumb base OA compared with an education control group.
Methods and analysis This is a randomised, controlled, single-centre, two-arm superiority trial with 1:1 allocation ratio; with assessor and statistician blinded. Participants are blinded to the trial's hypothesis and to the interventions received by the opposite group. A total of 204 participants will be recruited from the community and randomised using a computer-generated schedule. The intervention group will receive education for joint protection and OA, a splint for the base of the thumb, hand exercises and topical diclofenac sodium 1% gel over 6 weeks. The control group will receive education for joint protection and OA alone. Main inclusion criteria are pain ≥40 mm (Visual Analogue Scale, 0–100) at the base of the thumb, impairment in hand function ≥6 (Functional Index for Hand Osteoarthritis, 0–30) and radiographic thumb base OA (Kellgren Lawrence grade ≥2). Participants currently receiving any of the intervention components will be excluded. Outcomes will be measured at 2, 6 and 12 weeks. The primary outcome is change in pain and hand function from baseline to 6 weeks. Other outcomes include changes in grip and pinch strength, quality of life, presence of joint swelling and tenderness, duration of joint stiffness, patient's global assessment and use of rescue medication. Analysis will be performed according to the intention-to-treat principle. Adverse events will be monitored throughout the study.
Ethics and dissemination This protocol is approved by the local ethics committee (HREC/15/HAWKE/479). Dissemination will occur through presentations at international conferences and publication in peer-reviewed journals.
Trial registration number ACTRN12616000353493; Pre-results.
- osteoarthritis
- thumb
- conservative
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Strengths and limitations of this study
There is currently no highly efficacious strategy for managing thumb base osteoarthritis (OA). A combination of non-pharmacological and pharmacological modalities is frequently used in practice but evidence of efficacy for this strategy is lacking.
The 204 participants will be blinded to the treatment offered to the opposite group in order to minimise differences regarding expectations with the allocated treatment between groups.
Interventions included in this study are usually easily available in practice which facilitates implementation.
Results from the subgroup analysis will be used for hypothesis generation only, as the study was not powered for these analyses.
Introduction
Background and rationale
Osteoarthritis (OA) is a chronic and prevalent joint disorder with great impact on quality of life and high economic burden.1 ,2 The most common form of OA is that involving the hands, affecting three times more women than men and with greater prevalence following menopause.2 ,3 Overall prevalence of symptomatic hand OA is nearly 15%,4 while in the elderly (aged 70 years and over) the prevalence is about 26% in women and 13.4% in men.5
Among the different subtypes of hand OA, that affecting the base of the thumb represents a particular challenge to clinicians due to associated disabling symptoms and the limited efficacy of treatment options.6 Radiographic thumb base OA was reported to affect nearly 21% of the population over 40 years of age in the USA7 and it is usually more frequently related to pain and disability than interphalangeal joint OA.6 In addition to pain, it can cause deformity, stiffness, decreased range of motion and strength, resulting in difficulty performing common activities such as opening jars, carrying weights and writing.8
While thumb base OA is primarily treated with non-surgical modalities, surgical treatment may be indicated for those whose debilitating symptoms persist despite adequate conservative management. Thumb base OA is one of the most common causes of hand surgery.9 Surgical management, however, is associated with a number of complications, including tendon rupture, sensory changes and wound infection.10 In addition, a 7-year prospective study found 70% of patients waiting for surgery for thumb base OA were able to postpone or avoid surgery following conservative treatment with joint protection and splinting.11
Although a number of conservative therapies have proven to be effective for the management of hand OA, only modest treatment effects were reported for most individual interventions.12 This is particularly true in the case of thumb base OA, with few high-quality clinical trials in the literature to date and great heterogeneity across studies.13
Among the non-surgical modalities, education about the disease, delivered along with instructions regarding joint protection techniques, has been demonstrated to have no clinically significant benefits for pain.8 In contrast, a systematic review demonstrated that the use of splints for thumb base OA reduced pain, particularly in the long term, based on data from two trials.14 The same review found evidence that hand exercises might improve grip strength and hand function; however, evidence was based on individual trials using different exercise programmes. With regard to pharmacological management, there are insufficient data available to support the efficacy of intra-articular therapy with either corticosteroids or hyaluronic acid15 and their use is not recommended by the 2012 American College of Rheumatology (ACR) guidelines.16 On the other hand, non-steroidal anti-inflammatory drugs (NSAIDs) are recommended to relieve pain associated with thumb base OA, and topical formulations are recommended over oral NSAIDs in the most recent guidelines, due to a superior safety profile.16–18 However, their effect on function is small and transitory (no more effective than placebo after 2 weeks).19
Combining non-pharmacological and pharmacological modalities in the management of hand OA is recommended by the European League Against Rheumatism (EULAR) guidelines,17 supported by a literature review,8 and is frequently used in clinical practice. Despite this, direct evidence of efficacy of this strategy is lacking. A few trials evaluating combined treatment have been performed, although they have not generally been specific to thumb base OA.20 ,21 Moreover, the combinations investigated usually included exclusively non-pharmacological modalities22 and to date no strategy has been found to be highly efficacious for improving pain and function for thumb base OA. Determining an evidence-based therapeutic approach with a clinically meaningful effect on clinical outcomes would provide health professionals with a basis for decision-making for treatment of patients with thumb base OA. This strategy does not yet exist, and decisions on the best combination of interventions are usually based on personal experience and personal opinion of health professionals.
Objective
The aim of this study is to determine the effect of a combination of conservative non-pharmacological and pharmacological modalities in the treatment of thumb base OA compared with education control (standard care). The intervention group will consist of education about the disease and joint protection techniques, hand exercises, a splint to support the base of the thumb and topical NSAID. The control group will be provided with education about the disease and joint protection techniques alone.
Methods and analysis
Trial design
The COMBO trial is designed as a randomised, controlled, assessor and statistician blinded, parallel, two-arm superiority trial with 1:1 allocation ratio. The trial will be conducted at a single centre in Australia. The interventions will take place from baseline to 6 weeks and follow-up assessments will occur at 2, 6 and 12 weeks (figure 1). The protocol was designed in accordance with the principles of the Declaration of Helsinki.
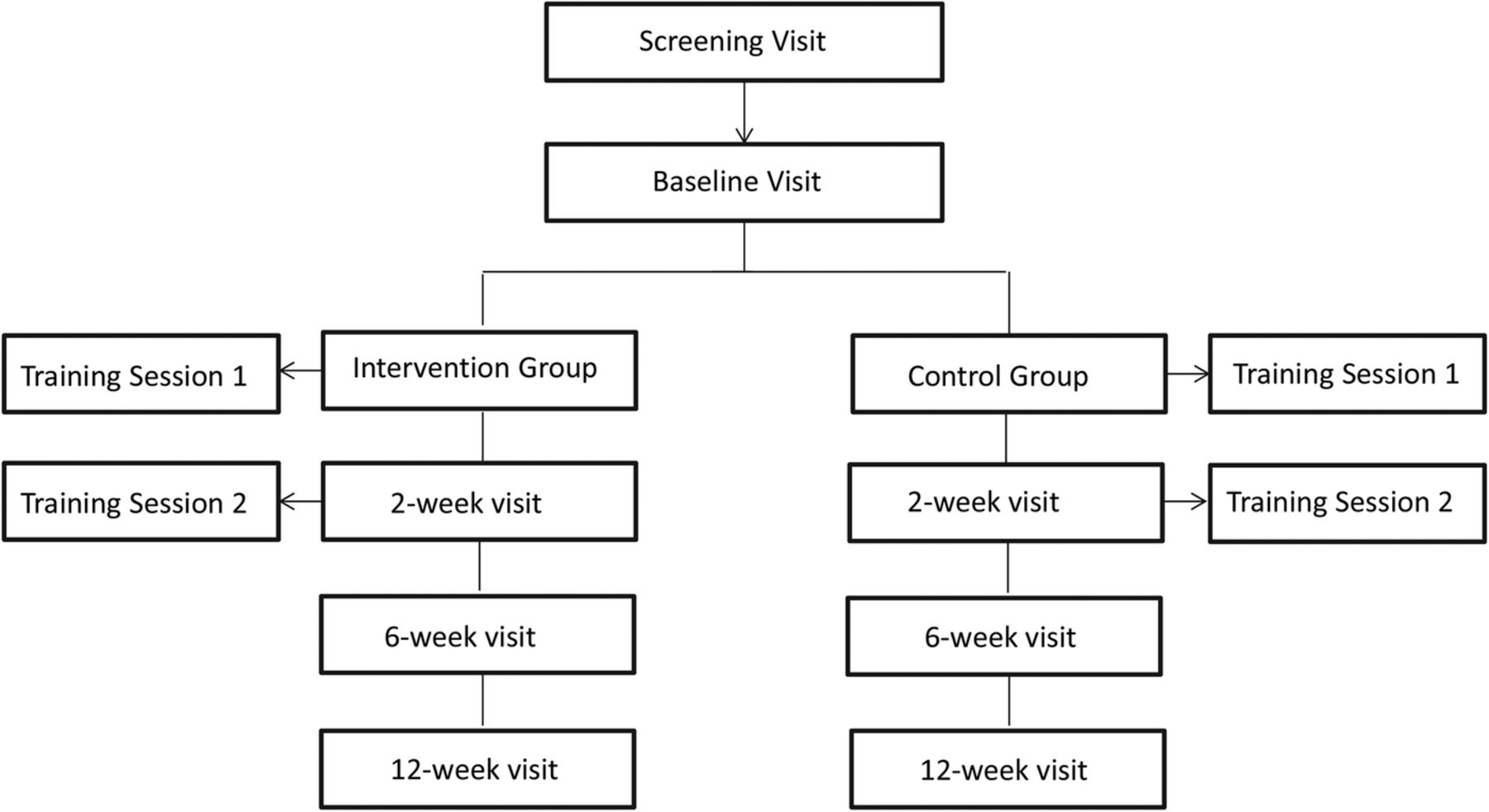
Trial design and visits.
Consumer involvement
Our consumer focus group has been involved in discussions during the genesis of this trial, which consisted of the selection of interventions, outcomes and the duration and frequency of the study assessments.
Participants
Participants will be recruited from the community and from our research volunteer database composed of participants who have expressed their willingness to be involved in future studies. The recruitment strategies will include advertisements on social media networks and posters/flyers placed on notice boards and waiting room walls of medical practices and community areas. In the first instance, a preliminary screening will occur by phone/internet and for those participants who pass this initial screening, a face-to-face visit will be conducted to confirm their eligibility. All participants will receive verbal and written information about the trial and the blinded assessor will obtain a written informed consent before inclusion.
Inclusion criteria
Participants will be eligible for the study if they meet all the following inclusion criteria:23 age ≥40 years; pain at the base of the thumb at least half of the days in the past month; average pain ≥40 on a 100 mm Visual Analogue Scale (VAS), where 0 is no pain and 100 is worst pain imaginable, over the past 30 days and in the 48 hours prior to the screening visit; scores ≥6 on the Functional Index for Hand Osteoarthritis (FIHOA, range 0–30)24 and radiographic evidence of thumb base OA read by a trained rheumatologist (Kellgren Lawrence grade (KLG) ≥2).25 The most severely involved hand will be included (as defined by pain VAS) in cases of bilateral symptomatic thumb base OA. If the pain scores are the same for both hands, participants will be asked to nominate the worst hand (ie, the one that causes more problems to the participant, either the dominant or non-dominant hand), which will be included as the index.
Exclusion criteria
Participants will be excluded if they fulfil any of the following criteria: known diagnosis of crystal-related arthritis (eg, gout, calcium pyrophosphate deposition disease), autoimmune arthritis (eg, rheumatoid arthritis, psoriatic arthritis), hemochromatosis or fibromyalgia; hand surgery in the last 6 months or planning to undergo surgery in the next 6 months; use of concomitant medications potentially directed at OA, unless at a stable dosage for at least 1 month for analgesics and NSAIDs or 3 months for slow-acting symptomatic or structure-modifying drugs (eg, diacerein, chondroitin sulfate, avocado/soybean unsaponifiables); intra-articular hyaluronic acid injection in the affected joint in the past 6 months; intra-articular steroid injection in the affected joint in the past month; significant injury to the affected joint in the past 6 months; any other self-reported hand condition that is likely to be contributing to the pain at the base of the thumb (eg, scaphoid fracture, carpal tunnel syndrome, DeQuervain's tendinopathy, trigger thumb, joint infection, diabetic neuropathy, pain referred from the neck, pain following hand or wrist trauma or surgery); poor general health likely to interfere with compliance or assessments, judged by the investigator; known hypersensitivity to diclofenac; current history of advanced renal failure; past or current history of gastrointestinal ulceration, bleeding and/or perforation; women who are pregnant or breastfeeding; current use of any of the study interventions.
Randomisation and allocation concealment
Individuals who consent to take part in the study and fulfil all study criteria will be assigned to either intervention or control group with a 1:1 allocation as per a computer-generated randomisation schedule, stratified by OA severity using KLG (2 and 3 vs 4) using random blocks of size 2, 4 and 6. The allocation sequence will be concealed from the researchers enrolling and assessing participants in sequentially numbered opaque, sealed and stapled envelopes. Aluminium foil inside the envelope will be used to render the envelope impermeable to intense light. Envelopes will be stored in a locked drawer and will be opened by the study coordinator only after the enrolled participant completes all baseline assessments.
The sequence generation will be prepared by a statistician and the envelopes will be prepared by an external investigator not involved in the trial. The study blinded assessor will enrol participants and the study therapist (one single physiotherapist with experience in thumb base OA management) will open the envelope in front of the participant and execute the designated intervention.
Blinding
All clinical assessments will be conducted by an assessor blinded to treatment allocation. To reduce the potential for unblinding, participants will be instructed not to disclose any information about the treatment, not to wear the splint during the follow-up assessments and not to use the topical NSAID just before the visit. Any occurrence of unblinding of the assessor will be recorded with its reason and reported along with the trial's results. The physiotherapist executing and supervising the treatments will not be blinded to the group allocation. The statistician involved in the main statistical analyses will be blinded to group allocation. Group allocation will be immediately unblinded if deemed necessary by the chief investigator in the case of serious adverse events potentially related to the study.
Participants will be naive to the treatments offered to the opposite group and hence blinded to the hypothesis of the study. In order to reduce the risk of bias, participants will be informed about the overall aspects involved in the treatment of both groups (ie, conservative therapies not involving oral or intra-articular medications) but not told the specific treatments included in each one of them nor about the differences among groups. This procedure aims to minimise differences regarding expectations with the allocated treatment between groups.
Interventions
The intervention group will receive a combination of education about OA and joint protection techniques, a splint to support the base of the thumb, hand exercises and topical NSAID. The control group will receive education about the disease and education about joint protection techniques alone (standard care). All participants will attend two individual, face-to-face treatment sessions with the study physiotherapist of ∼30 min each. The first will occur at the baseline visit and the second will occur after 2 weeks (2-week visit). Both treatment sessions will occur after the assessment by the blinded assessor. The 2-week visit aims at checking adherence to the programme while balancing the number of face-to-face visits and contact with the therapist between the control and intervention groups (two visits for each group).
The intervention will be delivered over 6 weeks. After the 6-week visit, participants will be encouraged by the physiotherapist to continue the treatment throughout the follow-up period (from 6 to 12 weeks) but intervention use will be at the participants' discretion. This period aims to evaluate participant's choice in continuing using the interventions and to assess the outcomes after this period.
Participants will be allowed to continue using current pain medications throughout the study provided the dose has been stable at study entry as per exclusion criteria. They will be permitted to use acetaminophen (paracetamol) as rescue medication for any symptom exacerbation, up to a maximum of 3000 mg/day.
Education about OA and joint protection techniques
All participants in this study (both groups) will be provided with education about OA and joint protection techniques through a nine-page educational booklet delivered at baseline (see online supplementary appendix 1) and through two face-to-face sessions with the study therapist at baseline and at the 2-week visit. Information about the disease will include anatomy of the first carpometacarpal (CMC) joint, using the Acland's video atlas of human anatomy (CMC joint of thumb),26 diagnosis, disease course, objectives of treatment, self-management and instruction on joint protection techniques and assistive devices. The booklet used in this study was adapted from an online resource produced by the hand therapy unit of James Cook University Hospital, UK.27 Written permission was obtained to use the adapted version. The physiotherapist will have a script to follow to maintain consistency with all participants regarding the joint protection advice.
supplementary appendix 1
Splint
While favourable results have been observed with different types of splints, prefabricated types are preferred over the custom-made version by most patients.13 Moreover, different ways of using the splint have been studied. A systematic review of design and effects of splints identified two trials with low risk of bias in which best results were achieved with the use of splints during daily activities.14 ,28 A prefabricated neoprene splint (Comfort Cool Thumb CMC Restriction Splint) will be used in this study. This splint is readily commercially available and does not require any customisation, facilitating potential dissemination post study, generalisability of our findings and application in routine clinical practice. The splint incorporates the base of the thumb and wrist and participants will receive recommendation to use it during activities of daily living (minimum of 4 hours/day) for 6 weeks (figure 2). The splint will be removed during rest, sleep, exercises and bathing.

{kind=link}
{kind=link}
Comfort Cool Thumb CMC Restriction Splint.
Hand exercises
The exercise programme will be the same for all participants in the intervention group and will consist of five exercises: thumb opposition, paper tearing, line tracing on ball, using chopsticks to pick up objects and squeezing a ball. The aim of the exercise programme will be to optimise range of motion (thumb opposition and line tracing on ball), to improve neuromuscular control of the alignment of the thumb and muscular endurance (paper tearing and squeezing a ball) and to train proprioception of the thumb base joint (all five exercises). Specific attention will be drawn to performing the exercises in a way which prevents collapse (hyperextension) of the first metacarpophalangeal (MCP) joint while maintaining the web space (abduction). These exercises are based on recent evidence which emphasises the importance of proprioceptive exercises and strengthening for the first dorsal interosseous muscles,29 ,30 while aiming to be functional and similar to movements used for daily tasks. The exercise programme will be visually depicted using images from a website developed by physiotherapists from the New South Wales Department of Health, Sydney, Australia (see online supplementary appendix 2).31 Consent was obtained to include the images into a written instruction to participants in the intervention group. The exercise programme was designed with emphasis on the first CMC joint in particular in order to optimise potential generalisability of the intervention for patients with this condition in clinical practice.
supplementary appendix 2
Participants will receive instructions on how to perform the exercises correctly during their supervised one-on-one session with the study physiotherapist at baseline. They will be further instructed to perform individual unsupervised at-home sessions, three times per week, from baseline to the 6-week visit and adjustment to the programme will be checked at the 2-week visit. Each exercise should be repeated 10 times during the first week. The muscular endurance exercises (paper tearing and squeezing a ball) will be progressed by increasing the difficulty of the tasks (ie, tearing a thicker paper and squeezing the ball harder (compressing the ball until it is about a third compressed (first week), half compressed (second week) and about three-quarters compressed (weeks 3–6)), respectively). For the remaining exercises, the number of repetitions will be increased, aiming for 12 repetitions during the second week and 15 repetitions for the following 4 weeks, if tolerated, as judged by the patient.
Topical NSAID
The intervention group will receive Diclofenac diethylammonium gel (11.6 mg/g) (diclofenac sodium 1% gel; Voltaren Emulgel), a topical NSAID commonly used in clinical practice that has been studied in large, good quality trials with superiority over placebo.18 Participants will be instructed to use the medication three times per day for 6 weeks on a daily basis. In order to standardise the amount used, participants will receive a small spatula with a permanent pen mark showing exactly how much product they should use. The advised amount corresponds with ∼200 mg to be applied in an area of 40 cm2, as recommended by the dosage guidelines.
Outcomes
Outcome measures used in this study are validated instruments that were promoted in recent recommendations for clinical trials for hand OA.23 The primary outcomes are change in pain scores at the base of the thumb, assessed by VAS (0–100 mm) and change in hand function, assessed by FIHOA (0–30) from baseline to 6 weeks. The FIHOA tool is composed of 10 items scored using a semiquantitative 4-point scale. It is a self-reported questionnaire evaluating the functional performance of 10 distinct activities involving the hand that has demonstrated good measurement properties, including reliability, feasibility and sensitivity to change.32
Secondary outcomes are change in pain scores at the base of thumb, assessed by VAS, and in hand function assessed by FIHOA from baseline to 2 and 12 weeks as well as the following outcomes assessed from baseline to 2, 6 and 12 weeks: change in grip strength (Jamar hand dynamometer (in kg)) and tip pinch strength (B&L pinch gauge (in kg)), assessed with participants with feet flat on the ground and elbow flexed at 90°; change in patient global disease assessment, assessed in response to the question ‘Considering all the ways your thumb arthritis affects you, how have you been during the last 48 hours?’ (on a VAS (0–100 mm, where 0 is very well and 100 is the very poor)); change in duration of thumb base stiffness, assessed by the question ‘What is the duration of stiffness at the base of your thumb in the morning?’ (expressed in minutes); change in health-related quality of life assessed by the Assessment of Quality of Life—4D instrument (AqoL-4D), a 12-item tool with good validity and reliability,33 including questions related to independent living, mental health, relationship and senses and scored from −0.04 to 1.00, with 1.00 indicating full health;34 use of rescue medications for pain at the base of the thumb (paracetamol up to 3000 mg/day), assessed by inspection of participant's diary; change in presence of swelling and tenderness, assessed by joint examination (scored as present or absent); participant's global rating of change for pain, function and overall change, assessed by the question ‘Which option best represents the change in pain/ change in function/overall change in your thumb since you began the study?’, scored using a 5-point Likert scale ranging from much better to much worse; percentage of treatment responders at 6 and 12 weeks according to the Outcome Measures in Rheumatology (OMERACT)-Osteoarthritis Research Society International (OARSI) criteria; and change in impairments in work and other activities, assessed by the Work Productivity and Activity Impairment Questionnaire-General Health (WPAI-GH), consisted of six questions related to impairments in work and daily activities over the past 7 days, which has been used in patients with rheumatoid arthritis and other rheumatic diseases.35–37
As tertiary/correlative outcomes, the participants will be required to fill out the credibility/expectancy questionnaire at baseline in order to assess expectation related to the assigned treatment.38 The questionnaire is composed of six questions and higher scores demonstrated higher expectation/credibility. In addition, range of motion of the first MCP joint will be measured with a goniometer and presence of first MCP joint collapse pattern on pinch will be recorded at baseline.
Procedures
An outline of the protocol visits and procedures is provided in table 1.
Schedule of study's events
Screening visit
After an initial screening phone call, potential participants will attend a screening visit that will consist of collection of demographic data such as age, date of birth, gender, ethnicity, financial status, marital status, symptom duration, current or past activities involving intensive use of the hands (eg, sports, gardening, playing specific musical instruments), previous and concomitant therapies, height, weight, years of formal education, primary occupation, comorbidities, menopausal status in women and OA at other joints (such as knee or hip). The key inclusion/exclusion criteria will be assessed to confirm the participant's eligibility.
Radiographic assessment
All potential participants will be referred for a posteroanterior (PA) view and an Eaton stress view radiograph of both hands. The protocol for acquisition of the Eaton stress view is presented in online supplementary appendix 3 and is based on a previous paper describing this technique.39 The PA view will be used to assess eligibility and thumb base OA severity using KLG, in addition to radiographic severity using the OARSI atlas40 and the Eaton classification criteria.41 The degree of subluxation of the first CMC joint will be assessed on the Eaton stress view.39
supplementary appendix 3
Baseline assessment
At the baseline visit, an ultrasound of the first CMC joint will be performed by a physiatrist with experience in musculoskeletal ultrasound, in order to assess presence of synovitis and other structural features, including osteophytes, cartilage damage, erosions and stability of the first CMC joint. The protocol for the ultrasound assessment is detailed in online supplementary appendix 4. Assessment of tenderness and swelling on physical examination by the blinded assessor will be first calibrated with the study's rheumatologist and inter-rater reliability will be assessed later in the study.
supplementary appendix 4
After the assessment, the blinded assessor will introduce the physiotherapist to the participant and leave the assessment room. At the end of the visit, the physiotherapist will deliver an envelope containing the credibility/expectancy questionnaire to assess the treatment expectation.38 The participant will be instructed to place the completed questionnaire back into the envelope and to seal it. The envelopes will not be opened until the study is complete.
2, 6 and 12-week visits
The outcome measures will be reassessed at the 2, 6 and 12-week visits by the blinded assessor. At the end of each visit, the study physiotherapist will record adverse events, use of rescue medication and treatment adherence from the participant diaries (see below). The physiotherapist will also ensure the correct performance of exercises by participants in the intervention group. Participants' global rating of change for pain, function and overall change will be concealed from the physiotherapist until the study is complete.
6-month contact with participants
After completion of the study at week 12, participants in the intervention group will be encouraged to continue the treatment and participants in the control group will be given advice about the interventions received by the treatment group (ie, hand exercises, splint and topical diclofenac), with the same recommendations as those used in the study for each intervention. All participants will be contacted by phone or through an online survey at 6 months to assess: (1) participants' choice in continue using the interventions (yes vs no); (2) the frequency of intervention use (as recommended by the physiotherapist vs less than the recommended by the physiotherapist); (3) primary outcomes (pain at the base of the thumb, assessed by VAS (0–100 mm); and hand function, assessed by FIHOA (0–30)). The reason for non-compliance (eg, lack of benefit, side effect, forgot to use intervention/do the exercise) will be recorded. This assessment aims at evaluating the effectiveness of the intervention in a longer term follow-up and will not be used for assessing between-group differences in treatment effects.
Treatment adherence
To monitor adherence to the treatments, participants from the intervention group will receive a diary and will be asked to record the hours of splint use and the use of topical NSAIDs on a daily basis. In addition, participants will be requested to report which exercises were performed and the frequency of these on a weekly basis. Adherence will be monitored using the diary from week 6 to 12, to assess whether participants continued the intervention following the 6-week visit. Control participants will also be provided with the diaries as all participants will be asked to record the use of rescue medication. Participants in the intervention group will be asked to record any adverse events relating to exercises, splint wear or use of topical NSAID in their diary.
Participant safety and withdrawal
Risk management and safety
Adverse events will be assessed by inspection of participants' diaries at 2, 6 and 12 weeks after intervention initiation. The risks for participants involved in this study are minimal. Topical NSAIDs have demonstrated a good safety profile and good tolerability, with no higher gastrointestinal, cardiovascular, renal and hepatic adverse events than placebo.18 Nevertheless, due to theoretical concerns related to the use of NSAIDs, adverse events will be monitored. To ensure the safety of participants, blood pressure will be monitored and systemic symptoms (new or worsening of previous symptoms) will be assessed at 2, 6 and 12 weeks after study initiation. Symptoms suspected to be related to the topical NSAIDs will be assessed by a physician involved in the study and, if necessary, the medication will be discontinued and further medical evaluation will be arranged. Local skin dermatitis may occur; however, these will most likely be minor local skin reactions. Participants who have local adverse reactions will cease the topical medication and will be followed to certify resolution of their rash. The same procedure will be applied if participants report local adverse skin reactions associated with the splint use. All adverse events will be reported.
Participants currently taking warfarin that are allocated to the intervention group will be required to consult their treating physician during the study due to the potential interaction between this drug and NSAIDs. Participants will be exposed to a small amount of radiation (0.001 mSV) for the acquisition of the radiograph, which is much lower than the annual average radiation dose (around 2 mSV).42
Handling of withdrawals
If a participant withdraws from the study, they will have their reasons for withdrawal recorded and any information provided or recorded up to the point of withdrawal will be kept in accordance with the data security and handling protocol of this study (see below). Strategies to maximise follow-up and prevent missing data will be used, including adhering to the assessment schedule in the event of participant withdrawal. In the event that the participant is unable to attend a study visit, the questionnaires will be administered over the phone. Participants who withdraw from the study will not be replaced.
Statistical methods
Sample size estimation and justification
The two primary outcome measures (VAS and FIHOA) were used to estimate the sample size. For the FIHOA, the minimal clinically important difference (MCID) is not known, thus the calculation was based on detecting a mean difference of 3 points (defined arbitrarily) on the FIHOA (range 0–30). The SD used was based on the baseline scores presented in the FIHOA's validation study (SD 6.2).43 For pain intensity, the calculation was based on detecting a MCID of 20 mm on a 100 mm VAS, assuming a SD of 20 mm as was used in a previous study.44 The two primary outcomes are correlated (r=0.49),24 hence an α of 0.027 was used as the level of significance for both outcomes, which preserves an overall 5% level of significance. To achieve a sample power of at least 80% for both outcomes, a sample size of 81 individuals per group will be required. To accommodate expected dropouts of 20% before study completion, we aim to include 102 participants in each group.
Statistical analysis
Data will be analysed according to the intention-to-treat principle. Demographic characteristics and baseline scores will be presented to assess comparability of treatment groups at baseline. Participants' characteristics will be described using mean and SD for continuous variables or medians (quartiles) if the distribution is skewed. Counts with percentages will be presented for categorical variables.
For continuous outcomes, the mean scores (SD) will be presented at each time point by treatment group. The between-group difference in mean change from baseline with 95% CI will be presented for all primary and secondary outcomes and compared using independent Student's t-test or the Wilcoxon rank sum test as appropriate. Categorical outcomes will be examined by χ2 test or Fisher's exact test, if expected cell counts are small. Analysis adjusted for baseline score and other relevant demographic and clinical characteristics will also be performed using analysis of covariance models fitted separately at 2, 6 and 12 weeks for all outcomes with the change from baseline as the dependent variable. Furthermore, standardised mean differences (95% CI) will be computed as the adjusted between-group difference in scores divided by the pooled SD of the baseline scores.
In addition, outcomes will be analysed on a categorical basis. The basis for categorisation will be the participant's score on the perceived ratings of change (participants reporting feeling much better and slightly better will be considered to have undergone meaningful change), and the OMERACT-OARSI criteria.45 Logistic regression models adjusted for age, gender, BMI and KLG will be used to compare response between treatment groups. The OMERACT-OARSI criteria for a meaningful change (improvement) are one of the following:
High improvement:
≥50% improvement+absolute change of ≥20 in self-reported pain intensity (VAS, 0–100 mm), or
≥50% improvement+absolute change of ≥6 in self-reported hand function (FIHOA, 0–30);
Or
Improvement in at least two of the three following:
≥20% improvement+absolute change ≥10 in self-reported pain intensity (VAS, 0–100 mm);
≥20% improvement+absolute change ≥10 in Patient Global Assessment of disease activity (VAS, 0–100 mm);
≥20% improvement+absolute change ≥3 in self-reported hand function (FIHOA, 0–30).
Post-hoc subgroup analyses will be performed examining whether there is heterogeneity in treatment effect according to presence of the following: concomitant symptomatic interphalangeal joint OA, presence of erosive hand OA (defined based on radiographic score), presence of CMC joint subluxation (assessed by the ratio of the radial subluxation of the base of the first metacarpal to the total articular width of the first metacarpal, on the Eaton stress view radiograph39) and by baseline OA severity according to KLG.
Data security and handling
Study data will be collected and managed using Research Electronic Data Capture (REDCap) tool hosted at the University of Sydney. This tool is a secure, web-based application designed to support data capture for research studies, providing (1) an intuitive interface for validated data entry; (2) audit trails for tracking data manipulation and export procedures; (3) automated export procedures for seamless data downloads to common statistical packages and (4) procedures for importing data from external sources. Backup reidentified information will be kept in password-protected electronic files. These files will be saved into an external hard drive that will be stored in a locked cabinet at the Principal Investigator's office. The privacy, security and ownership of the research data will be maintained and will not be stored or accessible by another organisation.
The archiving period for clinical research records will be 15 years. After this period, the electronic files will be deleted and paper forms will be destroyed. No information which could lead to the identification of a participant will be included in the dissemination of results.
Ethics and dissemination
Any protocol modification will be sent to review by the research ethics committee and will be amended at the trial registry. Dissemination is planned to occur through presentations at international conferences and publication in peer-reviewed journals.
References
Footnotes
Twitter Follow David Hunter @ProfDavidHunter and Bill Vicenzino @Bill_Vicenzino
Contributors LAD, DJH, AW, KLB, BV, PH, EAR, JPE, RJ and SRFM contributed to study conception and design. VD, ROC and WMO contributed to study design. DJH, KLB, BV, PH and SRFM attained project funding. LAD drafted the first version of the manuscript. ROC will have access to the final trial data set and perform the statistical analysis. All authors revised the protocol critically for important intellectual content and read and approved the final version of the protocol. All authors agree to be accountable for all aspects of the work.
Funding This work was supported by National Health and Medical Research Council (NHMRC) Program Grant (grant number APP1091302) and by the Lincoln Centre for Bone and Joint Diseases. KLB is supported by an NHMRC Principal Research Fellowship. DJH is supported by an NHMRC Practitioner Fellowship.
Disclaimer The funder had no role in the study design.
Competing interests DJH is a consultant to Flexion, Nestle and Merck Serono.
Ethics approval Local ethics committee Royal North Shore Hospital (HREC/15/HAWKE/479).
Provenance and peer review Not commissioned; externally peer reviewed.