Article Text

Abstract
Introduction Postcraniotomy pain protocols use opioids, which are considered suboptimal analgesia following this procedure. Multimodal analgesia components are sparse. Our null hypothesis states that sumatriptan is not different to placebo in addition to usual intravenous opioids, for the treatment of acute postcraniotomy pain.
Methods and analysis This is a prospective single-centre randomised double-blinded placebo-controlled phase III clinical trial comparing subcutaneous sumatriptan injection in the recovery area with placebo for the treatment of postcraniotomy pain. Eligible adult patients (18 years and older) undergoing craniotomy will be identified preoperatively. Both patient groups will receive a subcutaneous injection at a point where recovery-nursing staff would initiate the usual intravenous opioid analgesia as per standardised pain management protocol. In both groups, further pain management will be followed by the usual intravenous opioid administration. Primary outcome will consist of the difference in pain experienced by the two groups of patients in recovery area 60 min after the study drug administration. Postcraniotomy pain will be measured at regular intervals using the Visual Analogue Scale (VAS) in recovery area. The minimal clinically important difference of 10 mm on the VAS between the two groups will be considered as statistically significant. We will include selected clinical and patient-reported outcomes as secondary endpoints. Univariate regression will be conducted on each one of the clinically plausible potential confounders. We will enrol a total 136 patients, with the study duration of 2 years. This trial will commence recruitment on the 1 July 2019.
Ethics and dissemination This trial protocol has achieved approval by the Austin Health Research Committee, HREC/17/Austin/596. This trial was prospectively registered with Australian New Zealand Clinical Trials Registry on the 10/05/2018 with a unique trial identifier U1111-1209-9072 and registration Number ACTRN12618000793213P. Findings of this study will be disseminated in peer-reviewed academic journals.
Trial registration number U1111-1209-9072, ACTRN12618000793213P
- craniotomy
- postoperative pain
- analgesia
- sumatriptan
- Visual Analogue Scale
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
To our knowledge, subcutaneous sumatriptan use for treatment of postcraniotomy pain trial is the first randomised placebo-controlled double-blinded trial investigating the effectiveness of subcutaneous sumatriptan in the treatment of postcraniotomy pain.
This is a novel scientific hypothesis testing the utility of a ubiquitous antimigraine medication for a postoperative indication in this phase III clinical trial.
Ethical structure of the trial allowing for gold standard placebo use in analgesic therapy in addition to usual treatment, allowing for blinding of patients and investigators.
Utilisation of validated pain measurement scale in the form of Visual Analogue Scale, the timing of which has been tailored to fit the pharmacokinetic properties of injectable subcutaneous sumatriptan.
Involvement of hospital pharmacy in the randomisation sequence generation, with point of care allocation of randomisation envelopes allowing for blinding of investigators, patients and staff while maximising allocation efficiency.
Introduction
Postcraniotomy pain is often under-estimated and under-treated. Both acute and chronic postcraniotomy surgical pain and headaches have been found to be common and significant clinical phenomena.1 In a recent study by Mordhorst et al, 55% of patients had moderate to severe postoperative pain in the first 24 hours following craniotomy.2 In-hospital poorly controlled pain confers a significant morbid burden. It has been correlated with poor medium and long-term postoperative outcomes, including anxiety, depression, poor rehabilitation and development of chronic pain.3 Risk factors for increased acute postcraniotomy pain include female gender and surgical site of the incision. Opioids are still the mainstay of postoperative craniotomy pain management.4 Effective opioid analgesia administration for the purposes of postcraniotomy pain relief can reduce the clinician’s ability to monitor consciousness and result in decreased respiration with subsequent hypercarbia.
There is presently a limited scope for multimodal analgesia, due to lack of suitable medication components for this type of surgery.5 Ketamine and tramadol exhibit an unfavourable side-effect profile in relation to this type of surgery, with the adverse effect profile of both drugs including seizure risk. The use of non-steroidal anti-inflammatory agents has been restricted in neurosurgery due to their antiplatelet effects. In prior well-designed studies, intravenous parecoxib at skin closure was found to be ineffective at ameliorating postcraniotomy pain.6 Paracetamol has been found to modestly decrease postoperative pain scores but not the postoperative opioid consumption.7 There is a need for further clinical trials in order to improve and optimise multimodal postcraniotomy pain management in the short and longer term.2 8
Sumatriptan is a widely used drug, licensed for the treatment of migraines and cluster headache.9 10 There have been reports of its effectiveness for the treatment of medical conditions other than the ones already approved of by the relevant governing bodies. Sumatriptan has shown a promising therapeutic profile in patients suffering from trigeminal neuralgia in selected clinical studies.11 In a recent trial of sumatriptan use in minicraniotomy for decompression of trigeminal nerve, it was found to likely be as effective as the standard treatment modality when patient-reported outcome measures were evaluated.12 Sumatriptan use improved quality of recovery (QoR) scores 40 in patients undergoing mini-craniotomy for trigeminal nerve decompression.13 Further reviews have included sumatriptan in their reports of its effectiveness as a component of multimodal analgesia in the treatment of acute and chronic postcraniotomy pain.14 There are reports of the effectiveness of sumatriptan in analgesia regimens following vestibular schwannoma surgery.15
Sumatriptan is available in the oral immediate release form as well as the subcutaneous injection form. The medication penetrates the blood–brain barrier poorly, which is indicative of its peripheral mode of action. In terms of its pharmacodynamics profile, sumatriptan is a specific vascular 5-hydroxytryptamine-1B-D (5HT1B-D) receptor agonist with no effect at other 5HT receptor (5HT2-5HT7) subtypes.9 The vascular 5HT1 receptor is found predominantly in cranial blood vessels and mediates large cerebral artery and dural vessel vasoconstriction. Sumatriptan interacts with the trigeminovascular system in two distinct ways: through direct vessel constriction by its highly selective agonist activity at 5HT1B-D; it may also affect the modulation of the release of various inflammatory neuropeptides, including calcitoningene-related peptide (CGRP).11 16 CGRP is a proinflammatory neuropeptide released from trigeminal ganglia cells in migraine conditions.17 Pharmacotherapy with sumatriptan can both reduce CGRP release as well as the CGRP transcription. Prior studies have implicated CGRP in decreasing the proinflammatory state.18 Some of the newer studies have brought into question the exact mechanism of CGRP activity.17 Subcutaneous sumatriptan reaches its peak effect 6–20 min after administration. In controlled studies with sumatriptan injection, the most common adverse reaction with >2% risk of events, were injection site reactions, tingling, warm/hot sensations and burning sensation. Other very rare side effects include reports of adverse cardiac events as well as cerebrovascular events. In a number of cases, it appears that cerebrovascular events were primary. It is therefore very rare for sumatriptan to cause these complications with an incidence of <1%.
Immediate postcraniotomy pain is multifaceted, due to responses from injury of the skin, muscles and leptomeninges including the dura. The pain is usually described as a throbbing pulsating headache.4 Sources of postcraniotomy pain include tissue injury (scalp, cranial muscles soft tissue and dura mater) and nerve disruption, traction, entrapment and compression.19 The somatic component of the pain occurs due to the surgical incision and reflection of pericranial muscles and soft tissues of the scalp.4 Skull base surgeries employing suboccipital and subtemporal approaches produce higher degree of postoperative pain meningeal irritation also contributes to postsurgical pain. Nevertheless, it is the amount of tissue damage rather than the location of the surgery, which determines the intensity of postcraniotomy pain. Greater amount of tissue injury generates higher intensity of postoperative pain. Although the brain itself is not innervated, dura matter and the meninges, are rich in blood supply and pain receptors. Much of the postcraniotomy pain is contributed by the irritation of the dura and the meninges.4 We are hypothesising that in surgical cases of breaching the dura and leptomeninges, sumatriptan would exhibit the anti-CGRP-effect and therefore contribute to decreasing the activation of the trigeminovascular system.
Our null hypothesis states that sumatriptan is not different to placebo in addition to usual intravenous opioids, for the treatment of acute postcraniotomy pain. The alternative hypothesis states that sumatriptan is superior to placebo in addition to usual intravenous opioids, for the treatment of acute postcraniotomy pain. Our objective is to improve the available multimodal analgesic options for the treatment of postcraniotomy pain. Our primary outcome is centred around the measurement of postoperative pain score on Visual Analogue Scale (VAS: at 60 min). As a surrogate measurement of pain, we aim to measure the total opioids consumed and ancillary analgesics in both groups at similar points in time, in the recovery area and up to 24 hours postoperatively. We aim to measure satisfaction scores using QoR 40 scores at 24 hours.13 We will follow-up the patients at the intermediate time point of 30 days postoperatively.
Methods
Trial design
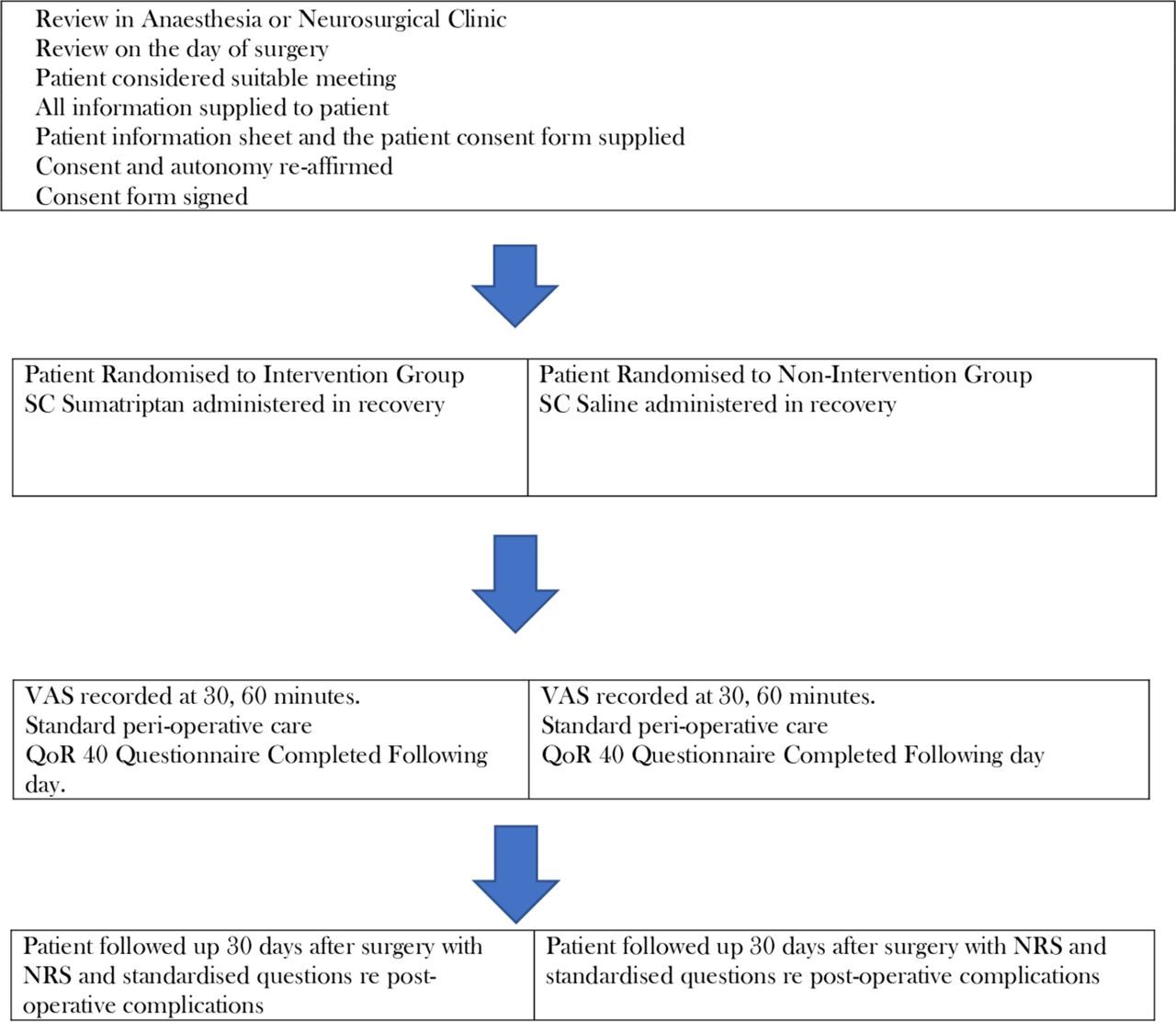
The sumatriptan for postcraniotomy pain clinical trial is designed as a randomised, placebo-controlled, anaesthetist, surgeon, patient and nurse blinded clinical study. This is a single-centre study undertaken at the Austin Health main hospital campus (as outlined in trial registration details). Study will be undertaken during the perioperative period. Process of recruitment will begin in the neurosurgical and anaesthesia clinics. Patient consent will be signed preoperatively, either in the preadmission clinic or in the preoperative area prior to delivering care in the preanaesthetic area. The actual intervention will be administered postoperatively in recovery during the pain management process. The primary endpoint of postoperative pain will be measured using the VAS. Randomisation will be performed as a block randomisation with a 1:1 ratio. The study drug will be administered at the usual point of the patient needing analgesia in recovery—when the patient complains of mild–moderate pain or gives a Numerical Rating Score (NRS) pain of at least 4/10. The initial subcutaneous sumatriptan injection will be compared with an initial placebo injection. The ongoing pain management would be standardised use of intravenous opioids as per recovery protocol in both groups. The Standard Protocol Items: Recommendations for Interventional Trials figure (figure 1) demonstrates the schedule of patient review, consent, enrolment, interventions and assessments in this trial. The timeline of patient involvement is illustrated in figure 2.

Standard Protocol Items: Recommendations for Interventional Trials figure. The schedule of enrolment interventions and assessments in the study. NRS,Numerical Rating Score; VAS, Visual Analogue Scale.
{kind=link}
{kind=link}
Participant timeline. NRS,Numerical Rating Score; VAS, Visual Analogue Scale.
Study registration
This protocol, patient information consent form as outlined in the PICF/person responsible PICF, as well as all other supporting documentation have been reviewed by the Austin Health Ethics Committee with respect to scientific content and compliance with applicable research and human subjects’ regulations. This trial protocol has achieved approval by the Austin Health Research Committee, reference HREC/17/Austin/596. This trial has been prospectively registered with Australian New Zealand Clinical Trials Registry with a unique trial identifier U1111-1209-9072.20 The principal investigators will make safety and progress reports to the HREC at the Austin Health at least annually and within 3 months of the study completion or termination.
Patient and public involvement
Patients, patient advisors and public were not involved in the development of the research question or in the design of the study. Patient involvement in the study includes completing the patient-reported outcome measures postoperatively in the form of questionnaires QoR 40. Results will be disseminated to participants at the completion of the study.
Inclusion and exclusion criteria
Patients who are at least 18 years old and who are undergoing craniotomy will be included in the study. Patients will need to be fully autonomous and able to give a valid consent for surgery and this particular study, or have mild underlying cognitive impairment only with the consent being given by the next of kin. Patients with any of the criteria listed in box 1 will be excluded.
Study exclusion criteria
Not autonomous, or have mild underlying cognitive impairment only, with the consent being withheld by the next of kin.
Craniotomy for cerebrovascular surgery (ie, cerebral aneurysm or arteriovenous malformation).
Previous ischaemic or haemorrhagic cerebrovascular accident (CVA).
Unstable angina or previous acute myocardial infarction (AMI).
Severe hepatic impairment.
Uncontrolled hypertension.
Previous sensitivity to sumatriptan.
Current treatment with monoamine oxidase inhibitors (MAOI’s).
Emergency re-do craniotomy.
The exclusion criteria for this trial have been designed to maximise patient safety, while accurately reflecting the available scientific body of knowledge on sumatriptan.9
Cerebral haemorrhage, subarachnoid haemorrhage, stroke and other cerebrovascular events have been reported in patients treated with subcutaneous sumatriptan, and some have resulted in fatalities.9 This may have occurred due to erroneous prescription of sumatriptan for non-migrainous conditions. Cerebrovascular surgery may very rarely result in adverse events such as cerebral haemorrhage or stroke. We undertook a further safety review in the light of this being a phase III clinical drug trial. We located one study demonstrating an average increase of 6±5 mm Hg in systolic arterial blood pressure, after administration of 100 mg of oral sumatriptan. The clinical significance of this finding in terms of potential adverse effects is uncertain.21 In healthy volunteers (N=18), a study evaluating the effects of sumatriptan on peripheral arterial reactivity failed to detect a clinically significant increase in peripheral resistance.9 In an initial large cohort study of 130 411 migraine sufferers by Velentgas et al, there was no association found between triptan use and risk of stroke.22 An increased overall risk of atypical stroke was found in the population prone to migraines, unrelated to any medication used.
The risk of stroke with the use of sumatriptan, both secondary to ischaemic or haemorrhagic cerebrovascular event is deemed to be and quoted at <1%. We have incorporated these quantifiable figures into our PICF. This information is identical to the level of risk, which is quoted in the Food and Drug Authority (FDA) prescribing information.9 As per FDA, sumatriptan in contraindicated in patients with cerebrovascular disorders, we have excluded the patients undergoing cerebrovascular surgery from participating in this trial.
Randomisation and study intervention
Blocked randomisation will be used to assign recruited participants to one of the two study groups—placebo or blinded therapeutic treatment group. Randomisation will be accomplished by the clinical research pharmacist using a sequence of computer-generated random numbers. Randomisation envelopes will be available at the point of care interface (operating theatre). Patients who actually require postoperative analgesic therapy to be administered will complete enrolment in the study at the point of care. This design has been chosen to maximise the efficiency of patient enrolment into the study. All preoperative and intraoperative care will be at the discretion of the treating team and will be in-line with the current best practice institutional principles for intracranial surgery. Participants will be randomly assigned to either control or experimental group with a 1:1 allocation ratio in permuted random blocks, as per a pharmacy generated randomisation schedule. Allocation concealment will be ensured, as the randomisation code will not be revealed until the patient is enrolled in the trial. From a scientific perspective, we do not plan to stratify our sample. We plan to deal with potential confounders through a univariate analysis and subsequent covariate multivariable logistic regression.
Randomisation envelopes will be made available only prior to the commencement of the study. Patients will be randomised at the point of care in order to maximise efficiency. The allocation sequence will be restricted and only available to the pharmacy randomisation staff, thereby ensuring the blinding of investigators. The implementation in the recovery stages will be completely independent of the randomisation group—and therefore both the assessor (recovery nursing staff) of the VAS scores and patients will be completely blinded. In the event of a report of a severe adverse event, Data Safety Monitoring Board (DSMB) will be notified and decision made on emergency un-blinding. Intervention in the group A, the group receiving the SC sumatriptan will be initiated by the recovery staff at the point at which they would normally give the intravenous opioid protocol for pain. Criteria for administration of the study drug would be equivalent to the criteria for the administration of the usual therapy of the recovery intravenous opioid analgesia: NRS indicating mild–moderate pain or a 4–6 pain on a scale from 0 to 10, as self-reported by the patient.23 It is at this point that the patient would be randomised in the study, and the randomisation envelope acquired. We have aimed to ensure the efficacy of trial enrolment, through point of care randomisation.
If the patient has been randomised to group A, the recovery nurse would be asked to collect the syringe from BOX A and administer the medication in a usual subcutaneous fashion. If the patient has been randomised to group B, the recovery nurse would be asked to collect the syringe from BOX B and administer the medication in a usual subcutaneous fashion. The relevant anaesthetic nurse will subsequently assess the NRS scores as per intravenous opioid protocol, that is, every 5 min. If the standard protocol criteria for opioid administration are met during the subsequent assessment, patient will receive the usual intravenous opioid protocol. All patients will receive usual high standard routine postoperative care. The only additional assessments in recovery area would be those using the VAS scores prior to drug administration, at 30 and 60 min poststudy drug administration. The dose selected for use in this trial by the principal investigators is 6 mg subcutaneously as a single injection. Our reasoning for choosing this dosing schedule includes that this is the recommended initial dose by the Australian Register of Therapeutic Goods. Most of the treatment effect is achieved from a single subcutaneous dose of sumatriptan.9 Although potentially therapeutically inferior, there is an established modality for the treatment of postsurgical pain in the form of opioids. Following the initial single-dose injection of subcutaneous sumatriptan, patients in both groups will be treated in the recovery area in a usual manner with an intravenous postoperative analgesia regime.
Criteria for discontinuation of the trial in individual patients include:
Persistent Glasgow Coma Score (GCS) <12 in recovery.
Significant surgical concern repotential intraoperative adverse features: potential intraoperative cerebrovascular accident.
Study protocol adherence reminders will be made on on-going basis with the recovery nursing staff. There will be regular brief monthly reminders at the nursing education sessions.
Study outcomes and their measures
Primary endpoints
Our primary objective is to determine if subcutaneous sumatriptan is superior to placebo, in addition to usual intravenous opioid in the management of postcraniotomy pain as measured using VAS 60 min after study drug administration. The primary outcome measure chosen was VAS at 60 min after placebo or subcutaneous Sumatriptan administration. From a pharmacokinetic perspective, the time from subcutaneous injection to peak concentration is 6–20 min. Due to the potential for postoperative impairment of cognitive function, affecting the accurate measurement of pain, VAS result at 60 min has been chosen as the primary outcome.
Secondary endpoints
VAS score at 30 min postsumatriptan administration has been chosen to coincide with the peak pharmacokinetic effect of sumatriptan postadministration (box 2). We will analyse the VAS at 30 min pain outcome, and compare and contrast this measure to our primary outcome.
Secondary endpoints
Visual Analogue Scale scores 30 min postoperatively.
Numerical Rating Scale (NRS) scores 24 hours postoperatively.
Total recovery area postoperative opioid consumption.
Total 24 hours postoperative opioid consumption.
Quality of recovery scores 40, 24 hours postoperatively (day 1).
Total hospital length of stay.
Patient satisfaction 30 days postoperatively.
NRS pain score 30 days postoperatively.
As a surrogate measure of pain, we will be assessing the total opioid consumption both in the recovery area and postoperatively at 24 hours. Patient satisfaction at the phone interview 30 days postoperatively will be measured with a simple yes or no binary outcome. Any potential adverse events will be documented at 30, 60 min and the following day on the data collection sheet. Data on all and any adverse events will be collected by the study investigators, and initially analysed qualitatively.
Sample size calculation
Our sample size calculation was based on the primary outcome and the significant difference of 10 mm basis points in pain measurement on the VAS. A recent article in British Journal of Anaesthesia outlined that Minimal Clinically Important Difference to patients is equivalent to 10 mm.24 We have therefore chosen this value as significant difference between the treatment and placebo group. We used STATA V.13 programme to calculate the sample size. With monitoring overall VAS scores in recovery postoperatively, the mean value was found to be 73 with a wide SD. In an article by Jones et al, the mean VAS scores in recovery for postcraniotomy patients, the VAS was found to be 34.25 With a range of conflicting research, the median point for VAS was determined to be 50 mm. We have defined the 10 mm difference in VAS scores between the two groups as clinically significant and statistically significant. If we observe a pain reduction of 10 mm down to 40 mm, we would therefore be likely to accept our scientific hypothesis and reject the null hypothesis (table 1).
Statistical measures
Due to the potential for loss to follow-up and missing data, we plan to enrol additional patients to a total of 136. Interim analysis will be conducted at the halfway point of the trial to assess for any differences between the groups. Unless there is overwhelming evidence with a difference in the effect of p<0.05, we plan to continue with the trial completion.
Statistical analysis plan
The intervention arm will be compared against the control for all primary analysis. Descriptive statistics (mean (SD) or median (IQR)) will be used for continuous variables. Normal data distribution will be confirmed through a histogram validation and Shapiro-Wilks test. We plan to use the Student’s t-test to compare the means of different groups for the continuous outcome of pain scores. Quantitative variables (continuous outcomes) will be compared using the Student’s t-test or Mann-Whitney U-test to compare independent means (table 2). When indicated, a one-way repeated measures Analysis of Variance (ANOVA) will be performed. Categorical variables will be presented as absolute frequencies and percentage and compared between the two groups using the χ2 or Fisher exact test. The OR will be calculated with its 95% CI for the categorical postoperative outcome variables. A Bonferroni correction will be applied for multiple comparisons. We will use the Bonferroni method to appropriately adjust the overall significance for multiple primary and secondary outcomes as needed.
Summary of methods of analysis for each variable
For subgroup analysis, we will use regression methods with appropriate interaction terms. Multivariable regression will be based on logistic regression for binary outcomes and linear regression for continuous outcomes. P values will be reported to four decimal places with p values <0.001 reported as p<0.001. STATA V.13r will be used for statistical analysis. For all tests, we will use two-sided p values with alpha <0.05 level of significance. There may be a number of patient-related or anaesthesia technique-related confounders, which may affect the outcome in this study (table 3). We will conduct a univariate regression analysis on the significance of each one of these parameters. With any of the above parameters demonstrating a two-tailed p value of <0.1, they would be entered in a multivariable regression model for each one of the primary and secondary outcomes. This strategy would be employed in order to assess any significant contribution of these factors on the primary and secondary outcomes of interest.
Potentially confounding clinical parameters
Data collected from all randomised participants regardless of protocol adherence will be assessed on an intention to treat basis and analysed accordingly. Therefore, any patients who have withdrawn or been lost to follow-up will be managed on an intention to treat basis. Should any patients withdraw, we will report reasons for doing so and compare the reasons qualitatively. Analysis of harms will be limited to participants who received the intervention.
Recruitment
The allocated time for this study is 24 months in order to meet the demands for the enrolment of 136 patients. We plan to inform the anaesthesia and neurosurgical clinics of the study where the initial patient contact is made. The research team will monitor the lists in advance and make early contact with eligible patients. Subsequently, the research team will meet the patients and discuss the informed consent. The subjects will be approached by both study investigators, and medical staff familiar with the study.
Data collection, management and analysis
Data will be collected from the standard anaesthesia chart. NRS will be documented as per protocol in the recovery charts. The VAS will be also be documented in the recovery section of the anaesthesia chart. QoR 40 questionnaires will be completed by patients the following day and filed in the notes by the attending nursing staff. Data on all adverse events during the first 24 hours will be collected. In the sumatriptan for postsurgical pain trial, all data will be entered electronically. Data will be entered in a coded de-identified manner with reidentifiable codes stored in separate file. This will be done at the Austin Health, where the data originated. Participant files are to be stored in numerical order and stored in a secure and accessible place and manner. Participant files will be maintained in digital password-protected storage for 15 years as required by regulation after completion of the study.
Safety monitoring
Our DSMB is independent of the study organisers. During the period of recruitment to the study, interim analysis after 50% recruitment will be supplied to the DSMB. DSMB will review whether the active intervention has been proven with the analysis of primary outcome and analyse the total number of adverse events documented. All adverse events occurring after entry into the study and until hospital discharge will be recorded. An adverse event that meets the criteria for a serious adverse event between study enrolment and hospital discharge will be reported to the local DSMB. DSMB will review the event(s) in an unbiased fashion and make an appropriate report to the sponsor (Austin Health) and Austin Health Human Research Ethics Committee. Life-threatening conditions (ie, immediate risk of death); severe or permanent disability, prolonged hospitalisation or a significant hazard will all be reportable to the DSMB.
Discussion
Significance
To our knowledge, the subcutaneous sumatriptan for the treatment of postcraniotomy pain trial is the first trial to evaluate the utility and effectiveness of subcutaneous sumatriptan as a component of multimodal analgesic regime for the management of postcraniotomy pain. Primary study outcome is measured using the tools that have good validity and reliability for measurement of pain. The advantages of VAS are that there is good evidence for responsiveness, validity and test–retest reliability.26 27 In studies attempting to validate the NRS, the VAS is used as the gold standard clinical measurement.28 The NRS is considered to have overall lower precision than VAS in the perioperative setting and it may therefore negatively bias the outcome of the study. In addition to this, the VAS is considered easier to administer in patients with any verbal difficulties.28
This study has been designed as a placebo controlled superiority trial. The trial would therefore not require the participants to forgo treatment they would otherwise receive. In this case, there are compelling methodological reasons to determine the efficacy of the intervention, and the patients who receive the initial placebo intervention will not be subject to any risk of serious or irreversible harm.29 Furthermore, the follow-up at a 30-day time-point may provide insight into the effects of study intervention on intermediate perception of pain. If the benefits proposed by our study are substantiated, this can have a significant impact on postcraniotomy multimodal analgesic patient care.
Limitations
Analgesic requirements are commonly used as post hoc measures of pain experience.28 whether this effectiveness of sumatriptan in reducing the opioid dosage at various end-points during the first 24 hours is addressed in the secondary outcomes. We will be recording any adverse events experienced by the patient in the course of the study. However, accurate opioid side effects may be difficult to define and measure secondary to potential confounding by other medical conditions and medications.
No stratification or matching will be performed during the recruitment and conduct stages of the trial. We have identified a significant number of potential clinical confounders. The most efficient and statistically feasible approach to dealing with a high number of potential confounders is in the analysis stages by conducting a univariate analysis. When each parameter demonstrates significance according to predetermined p level, it will be entered into a multivariable regression model for each primary and secondary outcome of interest. We plan to conduct three distinct subgroup analysis all with strong biological basis. Our study is underpowered to detect a statistically meaningful difference in these subgroups. However, our analysis will indicate an observed trend in the data.
Ethics and dissemination
The structure of our study necessitates that sometimes the study recruiters and investigators will also be the treating clinicians. Patients will always be given information that describes the proposed research as well as the form for withdrawal of consent (Supplementary file—Patient Information Consent Form-PICF 1.3).
Supplemental material
When reasonable to do so, patients will be invited back to the clinic to ask any further questions around the trial and consent process. A genuine scientific question has been posed which has the potential to improve future pain management in this group. Patients will be informed through a detailed consent process that they will not achieve any additional clinical care by participating in the study nor will they come to any harm by refusing to participate. The potential undue influence is therefore minimised through the principles of fully informed patient consent, equal care and clinical equipoise.29 There will be no additional invasive investigations occurring in the study participants, decreasing the risk of inconvenience and patient harm. Protecting potentially vulnerable adults with mild cognitive impairment, who are eligible to participate in the study is vitally important. The investigators believe that it is a scientific necessity to enrol this population in order to avoid selection bias in the study population. Decision-making capacity (DMC) of this subset of patients would have been evaluated in order to ensure validity of the surgical consent process. Equivalent DMC will be transferred to participation in the study.
Any modifications to the protocol which may impact the conduct of the study, the patient outcomes or have the ability to influence the safety of the patient, changes to the study objectives, study design or patient population will be communicated to the Austin Health Ethics Committee. Permission will be sought to modify the protocol prior to any significant changes.
Conclusion
Postcraniotomy pain management consists of opioids with limited multimodal analgesic therapeutic options. We have delineated a phase III clinical trial utilising a frequently administered antimigraine drug sumatriptan in its injectable subcutaneous form in the setting of postcraniotomy pain management. Subcutaneous sumatriptan use for treatment of postcraniotomy pain is a single-centre randomised double-blinded placebo-controlled superiority trial. Primary outcome is a VAS score rating 60 min after drug administration. Secondary outcomes consist of VAS rating 30 min following the study drug administration as well as total 24 hours postoperative opioid administration. With the design and conduct of this phase III clinical trial we intend to expand the evidence base of postcraniotomy analgesia management.
Trial status
This trial will be recruiting from the 1 July 2019. The trial is planned to run for 2 years. This trial protocol has achieved approval by the Austin Health Research Committee, reference HREC/17/Austin/596 (ethics approval—online supplementary file). This trial was prospectively registered with Australian New Zealand Clinical Trials Registry on the 10/05/2018.
Supplemental material
References
Footnotes
Contributors AL reviewed the scientific literature and contributed to the original protocol. JR contributed the original scientific hypothesis and the original protocol. AS contributed to the original protocol. XJ reviewed the scientific literature and contributed to the original protocol. JD reviewed the scientific literature and contributed to the manuscript.
Funding This trial has obtained internal funding from the Austin Neurosurgical Research Fund.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval 20180918 Letter HREC17 Austin596 New Study Ethics Approval.
Provenance and peer review Not commissioned; externally peer reviewed.