Article Text

Abstract
Introduction Rheumatoid arthritis (RA) and ankylosing spondylitis (AS) are chronic, systemic, inflammatory diseases, primarily in the musculoskeletal system. Pain and fatigue are key symptoms of RA and AS. Treatment presents a clinical challenge for several reasons, including the progressive nature of the diseases and the involvement of multiple pain mechanisms. Moreover, side effects of pain treatment pose an implicit risk. Currently, no well-controlled studies have investigated how medical cannabis affects pain and cognitive functions in RA and AS. The present study aims to evaluate the efficacy and safety of medical cannabis in the treatment of persistent pain in patients with RA and AS with low disease activity.
Methods and analysis A double-blinded, randomised, placebo-controlled study of cannabidiol (CBD), followed by an open label add-on of tetrahydrocannabinol (THC) with collection of clinical data and biological materials in RA and AS patients treated in routine care. The oral treatment with CBD in the experimental group is compared with placebo in a control group for 12 weeks, followed by an observational 12-week period with an open label add-on of THC in the primary CBD non-responders. Disease characteristics, psychological parameters, demographics, comorbidities, lifestyle factors, blood samples and serious adverse events are collected at baseline, after 12 and 24 weeks of treatment, and at a follow-up visit at 36 weeks. Data will be analysed in accordance with a predefined statistical analysis plan.
Ethics and dissemination The Danish Ethics Committee (S-20170217), the Danish Medicines Agency (S-2018010018) and the Danish Data Protection Agency approved the protocol. The project is registered in the European Clinical Trials Database (EudraCT 2017-004226-15). All participants will give written informed consent to participate prior to any study-related procedures. The results will be presented at international conferences and published in peer-reviewed journals.
- rheumatoid arthritis
- ankylosing spondylitis
- chronic pain
- treatment with medical cannabis
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
The randomised, double-blind and placebo-controlled design aims to determine outcome data (on the defined endpoints) and, thus, reduces the risk of bias, especially selection bias.
Recruitment in routine care is expected to appropriately reflect the patients and conditions in the two diagnostic groups.
The performance of a controlled study demands the use of medical cannabidiol, and tetrahydrocannabinol instead of plant extracts, that is, tea or herbal preparations.
There is no clinical evidence for the optimal dosage and application ranges. Thus, the treatment regimens for the drugs used are an extrapolation of expert knowledge.
Both primary and secondary endpoints are based on patient-reported outcome measurements and may be influenced by bias.
Introduction
The treatment of rheumatoid arthritis (RA) and ankylosing spondylitis (AS) has improved significantly over the last three decades.1–3 Chronic pain and fatigue are symptoms typical of these major inflammatory rheumatic disorders.4–6 Cognitive dysfunctions, such as concentration and memory problems, are also often reported in patients with chronic pain. These cognitive dysfunctions can be related to pain itself, sleep problems or reflect a side effect of the pharmacological treatment.4 6 RA affects the small joints of the hands and feet, but can also involve the larger joints.7 AS mainly affects the spinal and sacroiliac joints and is characterised by back pain and stiffness.8 Pain may involve nociceptive and non-nociceptive components and is based on the interaction between peripheral inflammation and central sensitisation.9 10 The immediate pain is triggered by the inflammation of the synovial tissue and/or consecutive oedema of the subchondral bone, and leads to a sensitisation of the peripheral nociceptors.11
Thus, chronic pain is likely to be due to peripheral joint and central neuropathic pain mechanisms at various stages.11–16
Treatment of moderate to severe chronic pain is difficult to overcome, for several reasons: heterogeneity of the patients in a given diagnostic group, the progressive nature of the disease, involvement of multiple pain mechanisms and the presence of comorbidities, particularly in elderly patients.17 The rheumatologist is likely to pay full attention to the anti-inflammatory treatment. This approach implies the fact that chronic pain associated with increased mortality can be overlooked.18 19
There is a lack of knowledge about the effect of cannabinoids in rheumatic diseases. Based on a Cochrane meta-analysis, the authors concluded that the existing clinical studies of c annabidiol (CBD) applied in monotherapy are of such poor quality that there is insufficient data to draw any conclusions about the effectiveness and/or long-term security of the compound.20
Currently, only very few studies have investigated how medical cannabis affects cognitive functions, such as concentration and attention.21 A few studies have investigated the impact of illegally obtained cannabis in RA.20 Furthermore, studies that have assessed medical cannabis did so mostly in the context of multiple sclerosis.20 22 23 In contrast to studies of recreational cannabis, the studies in persons with multiple sclerosis indicate that medical cannabis does not negatively affect cognition and could improve sleep quality. Given the limited data and the lack of a proper control condition, no definite conclusions of the potential cognitive impact of medical cannabis could be drawn.20 24
Hence, concerns about potential negative side effects of medical cannabis on cognition have led the Danish health authorities’ attention on a patient’s ability to drive safely.20 23 Furthermore, in the treatment of rheumatic diseases, there is no established routine nor rheumatological competence to prescribe medical cannabis. Consequently, there is considerable uncertainty and caution towards the use of medical cannabis, even in the North American countries, where it is already legal to prescribe these compounds for rheumatological conditions.23 24 This can lead to patients resorting to self-medication with cannabinoids.20 24 25 Thus, there is a strong need for high-quality studies of the efficacy and side effects of cannabinoids.
The overall aim of the study is to investigate the effect of medical cannabis on pain in patients with RA and AS, to elaborate on the potential dosage of CBD and tetrahydrocannabinol (THC) and to explore if and how the test compounds affect patients’ cognitive functions and sleep.
Materials and methods
Setting and study design
The study is an investigator-initiated, double-blinded, randomised, placebo-controlled intervention study of CBD, followed by an open label add-on of THC. It is designed to evaluate the efficacy and safety of medical cannabis, either as CBD or in the form of the combination treatment of CBD and THC as ‘add-on’ treatment for chronic pain in RA and AS. The patient-reported outcome measurement (PROM),26 a pain visual analogue scale (VAS) score27 at a value of at least 50 are the key inclusion criterion. The score range is from 0 to 100; a higher score indicates greater pain intensity. Thus, the null hypothesis, H0, is that receiving the active treatment with cannabis derivatives does not improve the pain situation in clinical assessment after 12, 24 and 36 weeks.
Clinical data and outcomes are registered in an electronic Case Report Form (eCRF), based on the Reuma-eCRF system available within the Danish nationwide registry DANBIO.15 28 DANBIO contains actualised data on ongoing treatment regiments, which therefore easily can be monitored.
Biological samples are collected via the Danish Rheumatologic Biobank.29 Patients are recruited from four Danish university hospital departments. Patient inclusion is planned to start in November 2018 and is expected to continue for 14 months.
Participants
The study population consists of the following:
Patients with seropositive RA1 currently treated with either conventional disease modifying antirheumatic drugs (cDMARDs) or biological disease modifying antirheumatic drugs (bDMARDs), and without clinical signs of arthritis, as assessed by a 40-swollen joint count.
Patients with AS, according to the modified New York criteria,2 currently receiving either non-steroidal anti-inflammatory drugs (NSAIDs) and/or bDMARD, who show an absence of clinical signs of axial and peripheral arthritis and enthesitis, and who have an Ankylosing Spondylitis Disease Activity Score (ASDAS) <2.1.
Inclusion criteria
Minimum pain VAS 50, both at screening and inclusion.
Disease duration ≥2 years.
Ongoing treatment or earlier attempt to treat with paracetamol or NSAIDs without clinical signs of arthritis or spondyloarthritis.
Analgesic treatment unchanged at least 4 weeks before trial start.
Exclusion criteria
Age <18 years.
Pregnancy, pregnancy wish or ongoing breast feeding.
C-reactive protein (CRP) >10 mg/L.
Comorbidities, more specific competitive rheumatological disorders, such as systemic lupus erythematosus, scleroderma, polymyositis or chronic pain condition based on a further clinical detectable aetiology (eg, fibromyalgia).
Evidence of serious uncontrolled concomitant cardiovascular, pneumological, neurological, endocrinological, gastroenterological, urogenital, nephrological or hepatic impairment.
Major surgery performed <8 weeks before randomisation or planned major surgical interventions.
Uncontrolled disease states, such as asthma, psoriasis or inflammatory bowel disease, where flares are commonly treated with oral or parenteral corticosteroids.
Evidence of active malignant disease, malignancies diagnosed or treated within the previous 2 years, including haematological malignancies and solid tumours.
Actual or previous harmful use of alcohol or drug abuse, in accordance with the WHO definition,30 within the previous 2 years.
Ongoing treatment with opioids and/or cannabis products and/or neuroleptics, or treatment terminated <4 weeks before inclusion.
Hypersensitivity to the study compounds.
Suspected for, or evidence of, active schizophrenia, other psychotic illness in the family history (first degree relatives), other significant psychiatric disorder or treated depression associated with underlying condition.
Epilepsy or recurrent seizures.
Use of strong cytochrome P450 3A4 inducers.
Experimental treatment
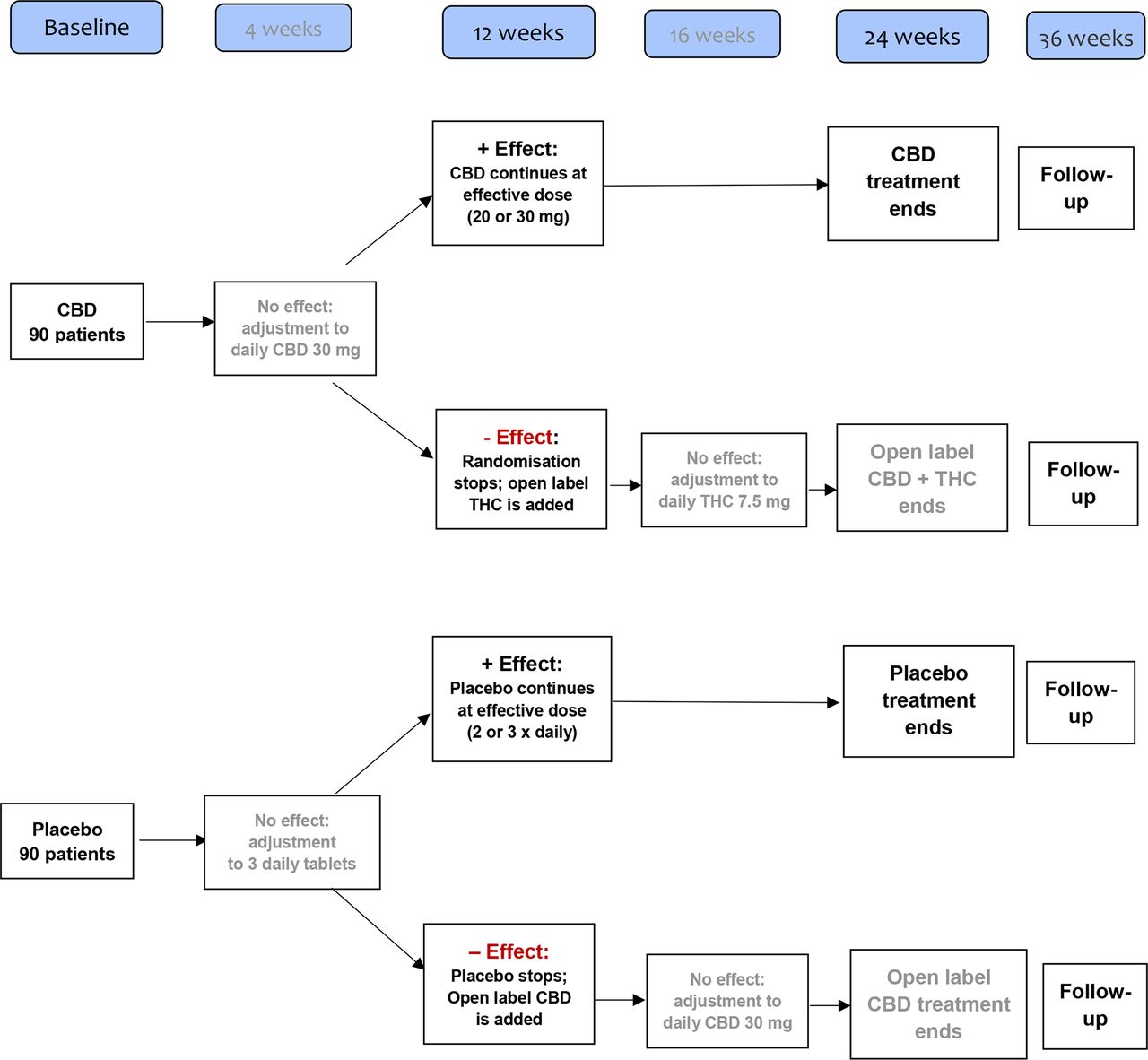
The treatment starts with oral CBD 10 mg or placebo before bedtime, and increases after 2 weeks to 10 mg two times per day. Finally, and in case of lack of effect (VAS-pain reduction <20) from the beginning of the fifth week, the treatment increases to 10 mg three times per day.
The clinical assessment after 12 weeks defines how to proceed during the following 12 weeks: in case of a sufficient response, that is, a VAS-pain reduction of ≥20, the established treatment continues randomised and without any further adjustment.
In case of insufficient response, that is, a VAS-pain reduction of <20, randomisation is terminated. Patients who received placebo are shifted to the active compound, that is, CBD treatment, and dose adjustment is performed, as mentioned above. In patients who received CBD treatment during the randomised period, the open label follow-up combines CBD with THC, that is, oral THC 2.5 mg daily is added to the ongoing CBD treatment.
The THC dose is increased after 2 weeks to 2.5 mg two times per day (in total, 5 mg THC/day), and in case of lack of effect (VAS-pain reduction <20, compared with VAS 20, as defined at clinical assessment after 12 weeks), after another 2 weeks to 2.5 mg three times per day (in total, 7.5 mg THC/day) from the beginning of the 17th week.
Figure 1 presents the consort flowchart and figure 2 the treatment flowchart.
Randomisation procedure
Patients are stratified by diagnosis and by recruiting centre. Patients are randomly allocated to one of the two treatment arms—CBD or Placebo—by random permuted blocks. Randomisation is blinded to the treatment allocation. Allocation is not known to anyone other than Glostrup Pharmacy, who produces and dispatches drug packages on request to each site. Sites receive a sealed, opaque envelope for each patient with the treatment allocation ready to be revealed, should this be required. Treatment is initiated within 2 weeks after randomisation. Measurements of effect are carried out at baseline before randomisation, and postintervention at 12, 24 and 36 weeks postrandomisation. Data analysis and statistical programming are blinded. The randomisation procedure and data analysis are performed by an independent statistician at the Department of Regional Health Research (IRS), University of Southern Denmark, Gråsten, Denmark.
Designated outcomes and clinical data
Primary outcome is the number of patients achieving an improvement of pain-VAS (Δ VAS-pain ≥20) after 12 weeks of treatment.
Secondary outcomes
The fraction (%) of RA and AS patients that achieve an improvement in VAS-pain, as assessed by the reduction of Δ VAS ≥20 and outcome of the PainDETECT Questionnaire,10 31 after 24 and 36 weeks.
The fraction (%) of RA and AS patients that achieve an improved quality of life (QoL) situation, as assessed by Global-VAS with Δ VAS reduction ≥20 and by the Short Form (36) Health Survey (SF-36),32 after 24 and 36 weeks.
The fraction (%) of AS patients that achieve a Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) <40 or reduction in BASDAI with Δ ≥20 after 12, 24 and 36 weeks.33
A characterisation of AS and RA patients’ cognition and sleep quality, as assessed by the Trail Making Test (TMT),34 the Digit Symbol Substitution Test (DSST)35 36 and the Pittsburgh Sleep Quality Index,37 performed at baseline and after 12, 24 and 36 weeks.
A characterisation of the patients’ expectation for the treatment effect, as assessed by the Credibility/Expectancy Questionnaire (CEQ) and by performing semistructured interviews38–42 at baseline and after 12 weeks.
A characterisation of and a final statement about serious adverse event (SAE) state.
The outcome measures include parameters recommended by the Initiative on Methods, Measurement, and Pain Assessment in Clinical Trials (IMMPACT) paper.43
Clinical data
At baseline, and after 12, 24 and 36 weeks, respectively, data are collected in the DANBIO Reuma-eCRF system. Furthermore, two additional nurse consultations are performed after 4 and 16 weeks, to obtain safety information and VAS-pain and to possibly perform a treatment increase from the beginning of the 5th and/or 17th week, respectively, as presented in the Experimental treatment section. The following data are collected at the time points as presented in figure 3.
Clinical measurements, that is, in RA the Disease Activity Score 28-joints (DAS28-CRP),44 45 Health Assessment Questionnaire and, in AS, the ASDAS and Bath Ankylosing Spondylitis scores for disease activity (BASDAI), function and measures are registered.46 47 In both patient groups, additional PROMs are obtained: VASs for pain, fatigue, patient’s global QoL score SF-36 and pain-score PainDETECT.10 31 Furthermore, the effect of intervention on attention and concentration is investigated using the TMT and DSST.34–36 Additionally, sleep quality is evaluated with the Pittsburgh Sleep Quality Index.37 The expected effect of treatment is measured with the CEQ and semistructured interviews.
Exposures, that is, all concomitant treatment, especially current treatments with cDMARDs, bDMARDs and/or analgesics, including dosing schedule and treatment onset.
Comorbidities, for example, cardiovascular disease, diabetes and hypertension.
Lifestyle (blood pressure, exercise habits and smoking status).
Patient demographics, for example, diagnosis, age, gender, height, weight, body mass index, disease duration, smoking status, educational level, marital status, sick leave, occupation and ethnicity are obtained at baseline.
Biological samples
Blood samples are obtained at baseline, and at 12, 24 and 36 weeks. In addition to routine blood tests, blood samples are collected in one EDTA tube (9 mL), two serum tubes (2×9 mL) and one PAXgene blood RNA tube (2.5 mL, Becton & Dickinson, Lyngby, Denmark), as described previously.48 These are collected for definition of drug concentration of CBD and THC, that is, monitoring of compliance, possible adverse events (AEs) and for further future analyses.
Statistical analysis plan
The power calculation is based on the following assumptions for the primary outcome.
An expected proportion with a response of 50% or more in the CBD group and expected 20% in the placebo group (OR=4). Response is defined as a reduction in VAS-pain of at least 20 (range 0–100) after 12 weeks of treatment.
A significance level of 0.05 in a two-sided z-test of proportions.
A total of 180 patients will be included in two balanced groups, each consisting of 90 patients.
This setup gives a statistical power of 0.98 for the primary outcome. The power is reduced if the true difference between the groups is less than the expected 30 percentage points, and if more than an expected 10% of the patients drop out of the experiment. Balanced groups of 45 patients will yield a power of 83% for two-group comparisons on binary outcomes, such as the primary outcome. Slight deviations in sample sizes might occur because of block randomisation.
The primary outcome is tested by a z-test in a logistic regression model. The main parameter estimates the ratio of the odds of response for the intervention group relative to the control group. All tests are two-sided. Secondary outcomes are analysed using logistic and linear regression, depending on the data type. In the case of deviations from the normality assumption, a non-parametric proportional odds model will be used. The secondary outcomes measured at baseline and postintervention, 12, 24 and 36 weeks (follow-up) will be analysed using mixed-effects models, controlling for time of measurement. The random-effects parameter is estimated for the clustering of repeated observations within patients. Analysis for the direct effects of CBD, THC and the interaction between those will be carried out separately, with placebo as the reference group for CBD.
Baseline measurements are reported as proportions of categorical variables, average and SD for normally distributed data, and median (range) scores for non-normal numerical data. All variables are reported for the two intervention groups. Baseline variables with a tendency (p<0.25) to coincide with the intervention group will be included as control variables in the test of the primary and secondary outcomes. The relationship between the tested variables at baseline and intervention allocation will be analysed with parametric (t-test) and non-parametric tests (χ2 and the Mann-Whitney test). Correction for multiple tests will be based on a gatekeeping model of access. This means that significant results for the secondary outcomes are interpreted solely as exploratory findings in case of a non-significant finding for the primary outcome.
Patient and public involvement
King Christian X’s Hospital for rheumatic diseases involves patients with inflammatory rheumatic diseases actively in both quality assurance projects, research projects and in the development of educational programmes. Furthermore, a user council was established in 2013 in the research department. The project is a consequence of rheumatic patient’s pain reality and the idea of the project was originally presented to the user council back in autumn 2017. Since then, two patients have been involved in the processing of the project. So far, the patient information brochures have been developed based on the integration of the patient’s perspectives. PROMs, especially the patient’s pain VAS, are the main focus of the outcome measurements. Thus, both the burden and consequence of the intervention and the results of the given intervention are transparent.
Meetings with the projects patient representatives will be arranged two times per year and the progress of the project is presented continuously for the user council.
Ethics and dissemination
All patients receive verbal and written information and give their written consent before enrolment, in accordance with Danish Ethics Committee guidelines. Online supplementary appendix 1 presents the projects consent statement in English. All patients are informed that they can withdraw from the study at any time. Although this would lead to the termination of project medication, patient withdrawal will have no consequences for regular course of treatment. In case of withdrawal, no subsequent patient-related registrations will be obtained.
Supplementary file 1
The two cannabis derivatives used in this study are comparable to the authorised compounds in the drug Sativex, which is a registered drug in DenmarK.49 The patients will receive the information that efficacy of the applied test compound, as well as potential side effect may be comparable to Sativex. The patient’s rheumatologist will provide relevant project information in an outpatient setting. Chronicity of the chosen diseases and the inclusion criteria implies the typical project patient to be well known with a serious burden of disease. Investigators and study nurses are specialists in the rheumatic field.
The treatment consists of CBD tablets and THC herbal capsule preparation, which are produced based on natural raw materials by Glostrup Pharmacy’s laboratory. The drugs are manufactured according to quality-ensured standardised procedures specifying the exact ingredients in milligrams. This makes dosage and monitoring of the therapy safe according to Danish national Good Clinical Practice (GCP) guidelines. The side effects, are well known and well described.50 The study subjects are patients who are already associated to one of the four participating outpatient clinics. The blood samples at baseline, after 12 and 24 weeks, respectively, will be realised in connection with routine blood tests, in accordance with an a priori arranged outpatient visit and thus will not pose increased risks. At all visits, participants will asked about events and/or reactions. Based on this information, the investigator will assess whether there is an AE, an adverse reaction, a SAE or a suspected unexpected serious adverse reaction. The GCP unit of the University of Southern Denmark monitors the study independently.
The patients will be contacted and informed regarding the overall study results if they indicate interest in this, and in accordance with the patient study consent form and as directed by the Danish Ethics Committee guidelines. The physician in charge of the project at each participating outpatient clinic is responsible for conducting the study in accordance with the fifth edition of the Helsinki declaration. Study participation does not affect the established anti-inflammatory treatment course of individual patients.
Results will be presented at international conferences and published in international and peer-reviewed medical journals. Negative, positive as well as inconclusive results will be published.
Discussion and potential limitations
The project’s focus is on chronic pain, which cannot be attributed to inflammatory activity.
Conventional and bDMARDs possess the potential to treat inflammation sufficiently. Thus, a treatment situation characterised by inflammatory disease activity should be treated according to the existing guidelines, that is, by adjusting the treatment to an adequate DMARD regiment.1 2 Consequently, the absence of inflammation is a major inclusion criteria. Potential participants are well known as the study demands that their course of treatment has taken place for at least 2 years. Thus, we assume that alcohol or drug abuse, as well as information about ongoing opioid and/or cannabis treatment are accessible information for the involved investigators. Furthermore, the demand of a disease duration of at least 2 years is supposed to ensure the presence of chronical pain.
CBD and THC are two of >80 active compounds in the marijuana plant.51
In contrast to THC, CBD does not exhibit a narcotic effect and/or intoxication.52 53 The biochemical effect of the cannabinoids is explained by the compounds’ interaction with specific receptors; the Cannabinoid receptor 1 (CB-1) receptor is located on neurons and glial cells in different parts of the central nervous system, whereas the Cannabinoid receptor 2 (CB-2) receptor is found in structures of the immune system. The stimulating and narcotic effects of THC are considered to be caused by activation of CB-1 receptors. CBD has a very low affinity for these receptors.51 Thus, CBD binding to the CB-1 receptors causes little to no narcotic effect. New studies show evidence that CBD affects autoimmune signalling pathways and that these mechanisms may be relevant to CBD’s therapeutic profile.52 53
The effect of CBD is studied in a placebo-controlled design, whereas the effect of a combination of CBD and THC is an open label continuation of the study. The scientifically ideal solution would have been a randomised study comparing both CBD, THC and placebo, for instance, in a cross-over design. Such a design would be characterised by the implicit risks of THC for all patients during the entire study period and it would require a significantly larger study. The actual design represents the balance between a wish to assess the effect of both CBD and THC correctly, while recognising risks, including traffic safety issues, especially due to the THC treatment. Also, the possible negative effect on cognitive functioning can have a large impact on job functioning. Therefore, a more definite answer as to whether medical cannabis negatively affects cognition is important in relation to job functioning and autonomy. We feel our design will provide important information on THC, despite the design, and it has the advantage that we know when THC is applied, and thereby can take the necessary precautions.
The trial population is monitored regularly at the participating outpatient clinics and the individual longitudinal treatment is registered. DANBIO is the nationwide clinical quality database for rheumatology.16 29 All adult patients treated with biological drugs are registered. Furthermore, patients with AS and RA are registered, regardless of treatment. Thus, the DANBIO based Reuma-eCRF system provides particularly good conditions for the collection and monitoring of validated data.

Presents the consort flowchart. TMT, Trail Making Test; VAS, visual analogue scale.

Presents the treatment flowchart. CBD, cannabidiol; THC, tetrahydrocannabinol.

{kind=link}
{kind=link}
{kind=link}
The schedule of assessments and procedures. AE, adverse event; ASDAS, Ankylosing Spondylitis Disease Activity Score; BAS, Bath Ankylosing Spondylitis; BP, bloodpressure; CEQ, Credibility/Expectancy Questionnaire; DAS, Disease Activity Score; DSST, Digit Symbol Substitution Test; SAE, serious adverse event; TMT, Trail Making Test; VAS, visual analogue scale.
References
Footnotes
Contributors OH wrote the protocol for the European and the Danish Medicines Agency and the Research Ethics Committee. TEA, AAC, JP, EMH, TE, TIH, AGB, AGL, ABB, MØ, MLH, NSK, KKR, KHP and OH contributed to study concept. KHP, MØ and OH drafted and revised the manuscript after feedback from all authors. All authors contributed to the review of the present manuscript and approved the final version of the manuscript.
Funding The Danish Rheumatism Association, represented by OH. The Danish Rheumatism Association, The Region of Southern Denmark and the Knud and Edith Eriksen Foundation have funded this investigator-initiated study.
Disclaimer OH: has received fees for speaking and/or and travel expenses from AbbVie, Roche, Novartis and Pfizer. JP: has received speaker and/or consulting fees and refund for travelling expenses from Pfizer and BMS. EMH: has received fees for speaking and/or consulting from MSD, AbbVie, UCB and Sobi; research funding to Aarhus University Hospital from Roche and Novartis; and travel expenses from Pfizer, Celgene, MSD, Sobi and Roche. AGL: has received fees for speaking and/or consulting from MSD, Novartis and Pfizer; research funding from Novartis and travel expenses from AbbVie, MSD, Novartis, Pfizer and Roche. MØ: has received speaker and/or consulting fees and/or travel expenses from AbbVie, BMS, Boehringer-Ingelheim, Celgene, Eli-Lilly, Hospira, Janssen, Merck, Novartis, Novo, Orion, Pfizer, Regeneron, Roche and UCB, and research support from Abbvie, Celgene, Centocor, Merck and Novartis. MLH: has received fees for speeking and or consulting from AbbVie, Biogen, BrMS, CellTrion, Eli Lilly, MSD, Novartis, Orion, Pfizer, Roche and Samsung. KHP: has received travel expenses from Roche and Pfizer
Competing interests None declared.
Ethics approval The protocol (version 8.1., 22 November 2018) is approved by the Danish Ethics Committee (S-20170217), the Danish Data Protection Agency (2018-41-5388) and the Danish Medicines Agency (2018-0 10 018).
Provenance and peer review Not commissioned; externally peer reviewed.
Patient consent for publication Not required.