Article Text

Abstract
Introduction After deep vein thrombosis, up to 50% of patients develop post-thrombotic syndrome (PTS). PTS is a chronic condition that reduces quality of life (QOL). Cornerstones of PTS treatment include the use of elastic compression stockings but this treatment is usually incompletely effective and is burdensome. Venoactive drugs have been reported to be effective to treat chronic venous insufficiency (CVI). However, the level of evidence supporting their use in CVI in general and in PTS in particular is low.
Methods and analysis The MUFFIN-PTS trial is an academic, publically funded, multicentre randomised placebo-controlled trial assessing the efficacy of micronised purified flavonoid fraction (MPFF, Venixxa), a venoactive drug, to treat PTS. Eighty-six patients with PTS (Villalta score (VS) ≥5) and experiencing at least two of the following PTS manifestations among daily leg heaviness, cramps, pain or oedema will be randomised to receive 1000 mg of oral MPFF or a similar appearing placebo for 6 months, in addition to their usual PTS treatment. Total study follow-up will be 9 months, with visits at inclusion/baseline, 3, 6 and 9 months. Primary outcome is the proportion of patients with improvement in VS in each group, where improvement is defined as a decrease of at least 30% in VS or a VS <5 in the PTS-affected leg. Main secondary outcomes include QOL and patient satisfaction.
Ethics and dissemination Primary ethics approval was received from Centre intégré universitaire de santé et de services sociaux (CIUSSS) West-Central Montreal Research Ethics Board. Results of the study will be disseminated via peer-reviewed publications and presentations at scientific conferences.
Trial registration number ClinicalTrials.gov Registry (NCT03833024); Pre-results.
- clinical trials
- thromboembolism
- vascular medicine
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
Multicentre, double-blind and publicly funded study.
Conducted by pan-Canadian CanVECTOR, patient-oriented research network dedicated to venous thromboembolism with a proven track record of completing high-impact multicentre clinical trials.
Results of MUFFIN-PTS trial will be highly relevant for both patients and healthcare providers and may change the clinical management of post-thrombotic syndrome.
Treatment duration (6 months) might be perceived as too short.
If trial is positive, another trial with longer follow-up may be necessary to assess long-term efficacy and side effects of micronised purified flavonoid fraction.
Introduction
The post-thrombotic syndrome (PTS) refers to the clinical manifestations of chronic venous insufficiency (CVI) that occur after a deep vein thrombosis (DVT).1 PTS is the most frequent complication of DVT.2 Despite treatment of DVT with anticoagulants, PTS occurs in up to 50% of patients after proximal DVT, and 5%–10% of patients with DVT develop severe PTS.3
While not life-threatening, PTS constitutes an important health problem as it reduces quality of life (QOL) and is associated with increased costs and health resource utilisation.4 The current cornerstone of PTS treatment is the use of elastic compression stockings (ECS).5 However, ECS are often not fully effective as judged by clinicians6 7 and not perceived to be effective in about one-third of patients.8 9 More importantly, use of ECS is burdensome as they must be worn each day from morning to evening,1 which limits compliance to ECS and, ultimately, ECS effectiveness.9
Thus, there is a pressing need for new therapeutic targets for the treatment of PTS.1 10–13 Venoactive drugs or phlebotonics which comprise a heterogeneous group of medicinal products of plant or synthetic origin could constitute an attractive option.14 They are simple-to-use oral medications, seem to be well tolerated and are potentially better pathophysiologically targeted than ECS (Li et al, Micronized purified flavonoid fraction for the treatment of chronic venous insufficiency, with a focus on post-thrombotic syndrome: A narrative review (under review)). Venoactive drugs have shown some effectiveness in patients with CVI14 and are suggested as a second option, after ECS failure, to treat swelling and pain related to CVI in the latest European Society of Vascular Surgery guidelines (grade IIa).15 However, venoactive drugs have not been definitively evaluated for the treatment of PTS and their effectiveness in this indication is unknown. Thus, international guidelines on the management of venous thromboembolic (VTE) disease currently do not recommend or even suggest their use to treat PTS.5 12 16 A well-conducted randomised controlled trial (RCT) assessing venoactive drugs to treat PTS is therefore needed.
Methods and analysis
The Micronized Purified Flavonoid Fraction (MPFF) for the Treatment of Post-Thrombotic Syndrome trial (MUFFIN-PTS) is a double-blind multicentre RCT assessing the efficacy of a 6-month course of MPFF versus placebo in patients with PTS.
The venoactive drug used in this trial is MPFF (commercialised under the name of Venixxa in Canada and Daflon/Venarus in some other countries). MPFF contains 90% micronised diosmin and 10% of flavonoids expressed as hesperidin.17 The micronisation of the drug to particles with a diameter <2 µm improves MPFF oral absorption, shortens its onset of action and potentially improves its efficacy in relieving symptoms and signs of CVI.18 19 Studies and literature reviews suggest that MPFF is effective to treat CVI,14 20 21 might be effective to treat PTS14 20 22 23 and is safe.20
Objectives of the trial
The primary objective of the MUFFIN-PTS trial is to assess whether a 6-month course of 500 mg two times per day of MPFF (Venixxa) improves PTS as compared with a placebo drug.
Secondary objectives are to assess:
If MPFF improves PTS severity as compared with placebo.
If MPFF improves each individual component of the Villalta score (VS) as compared with placebo.
If MPFF improves venous disease-specific QOL and generic QOL as compared with placebo.
If MPFF improves patient satisfaction with treatment as compared with placebo.
Patient compliance with treatment.
Serious adverse events (SAEs) including drug-related SAE, DVT, pulmonary embolism (PE) and death.
If MPFF is cost-effective. This will be assessed only if MPFF is found to be superior to placebo in the primary outcome analysis.
Description of trial procedures
This study is being conducted in seven Canadian centres. The clinical trial coordinating centre is located in Montreal (Lady Davis Institute, Jewish General Hospital). Co-principal investigators are Drs Jean-Philippe Galanaud and Susan R Kahn. All investigators are members of the Canadian Institute of Health Research (CIHR)-funded CanVECTOR research network. Patients with PTS (VS ≥5), as per International Society on Thrombosis Haemostasis (ISTH) guidance for PTS diagnosis,24 and at least two of the following daily PTS manifestations (leg heaviness, cramps, pain, oedema) will be potentially eligible to participate. The study inclusion and exclusion criteria as well as timeline and procedures are summarised in figure 1. Eligible and consenting participants will be randomised to receive either MPFF (Venixxa) 500 mg oral or oral placebo capsules identical in appearance to MPFF, 1 capsule two times per day (morning and evening) for 6 months. Placebo will be manufactured by Bay Area Research Logistics (Hamilton, Ontario, Canada), a Health Canada-approved company, and will look identical to MPFF/Venixxa capsules. Study drugs will be packaged in identical plain boxes, labelled with a unique code for that participant (ie, patients and study personnel blinded). Randomisation will be done online via a web-based system (Information Management Services (IMS), Montreal, Quebec, Canada). Patients will be allowed to continue all other previous treatments for PTS, except venoactive drugs (which are very rarely used in Canada). This includes the use of ECS, exercise training and lifestyle measures (eg, leg elevation, avoiding a sedentary lifestyle, avoiding prolonged exposure to heat, maintaining a non-obese body weight and using moisturising creams to avoid skin breakdown).16 Standard treatment of PTS (ECS, exercise training, lifestyle changes, or rarely, interventional radiological procedures for severe PTS) is expected to be similar at all study sites across Canada and is expected to be balanced between groups. Information on ECS use, strength and compliance will be documented at baseline and each follow-up visit as ECS could impact assessment of our primary outcome.
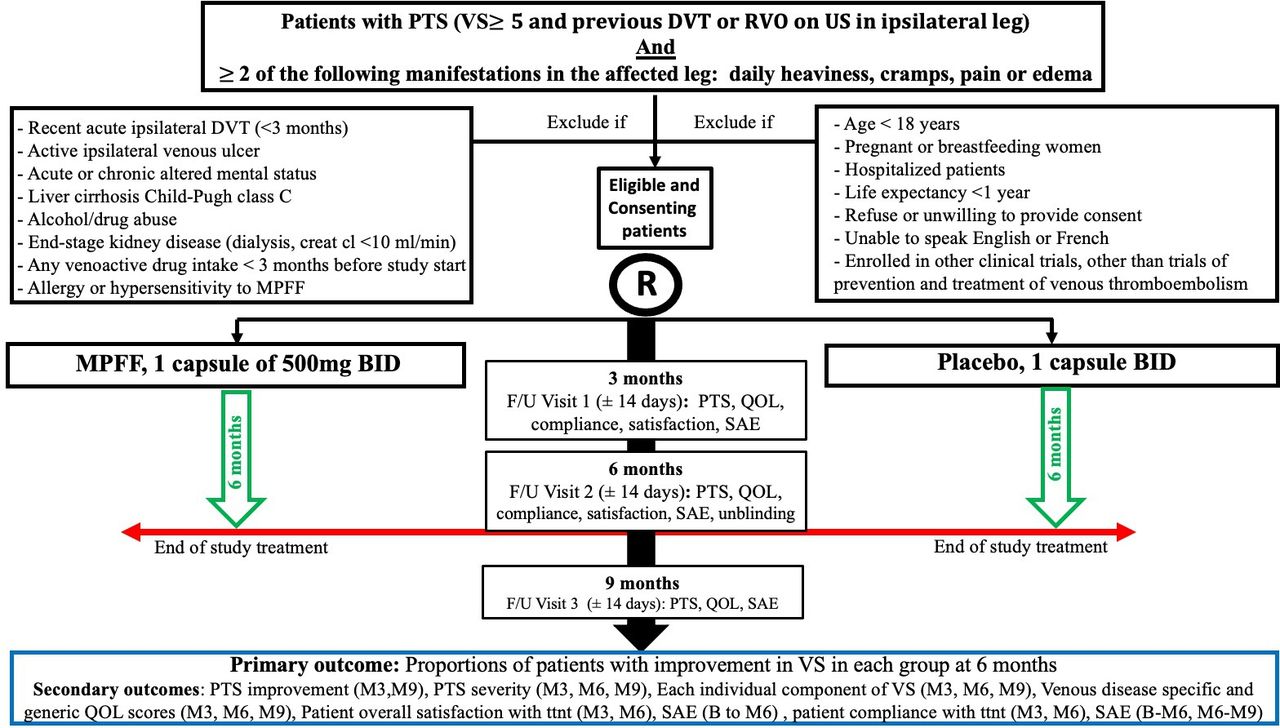
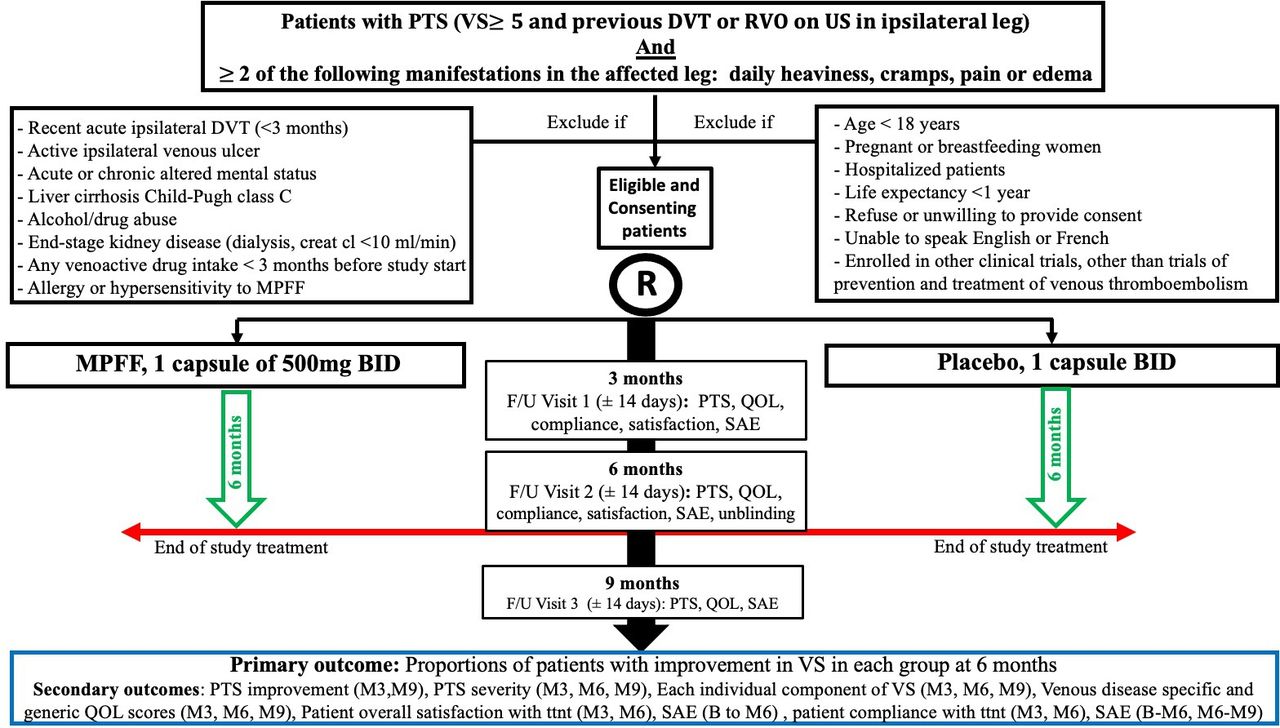{kind=link}
MUFFIN-PTS flow diagram. B, baseline; BID, two times per day; creat cl, creatinine clearance; DVT, deep vein thrombosis; F/U, follow-up; M3, 3-month F/U visit; M6, 6-month F/U visit; M9, 9-month F/U visit; MPFF, micronised purified flavonoid fraction; PTS, post-thrombotic syndrome; QOL, quality of life; RVO, residual venous obstruction; SAE, serious adverse event; US, ultrasound; VS, Villalta score.
Study participants will be followed for 9 months after enrolment. They will have scheduled clinical assessments at baseline (day 0: day of enrolment), 3, 6 and 9 months (±14 days). The objective of the 9-month follow-up visit is to assess if any between-group differences noted at 6 months have changed by 3 months after the end of study drug treatment.
Unscheduled assessments will take place if patients have symptoms suggestive of a SAE. Details of study procedure for each follow-up visit are presented in table 1. If a patient is unable to attend any of the follow-up visits in person, a telephone follow-up visit will be scheduled to assess PTS using the validated patient-reported Villalta scale.25 However, in-person attendance at all follow-up visits will be strongly encouraged. In the event a patient discontinues study drug, he/she will be asked to complete all study follow-up visits.
MUFFIN-PTS trial: procedures undertaken at each follow-up visit
Assessment of outcomes
Primary outcome
Improvement of PTS at 6 months. As recommended by the ISTH, PTS will be assessed with the Villalta scale in the DVT-affected leg.24 In the case of bilateral PTS, we will use the leg with the highest Villalta score at inclusion for assessment of efficacy of MPFF; for patients with unilateral PTS and a VS ≥5 in the leg contralateral to DVT (ie, non-PTS-related venous insufficiency), we will use the DVT-affected leg for assessment of study drug efficacy. Improvement will be defined by: absence of PTS at the 6-month visit (VS <5 in the PTS-affected leg) or, in patients who still have PTS, a decrease of at least 30% in the VS between baseline and 6-month assessments.26 Improvement (reduction) in VS rather than complete resolution of symptoms and signs of PTS was chosen to assess the efficacy of MPFF because (1) some patients may have persistent/irreversible symptoms and signs of PTS such as hyperpigmentation or lipodermatosclerosis which can prevent them from ever having a VS <5 during study follow-up; and (2) given the current absence of effective treatments for PTS, even incomplete relief of symptoms and signs is desirable.
Secondary outcomes
Severity of PTS at 3, 6 and 9 months will be assessed (1) based on the validated Villalta scale severity categories, namely: mild, VS 5–9; moderate, 10–14; and severe, >14 or presence of ulcer; and (2) based on the VS (continuous variable).24
QOL at 3, 6 and 9 months: will be measured using validated generic (EuroQol 5-Dimension 5-Level (EQ-5D-5L) questionnaire) and venous disease-specific (VEINES-QOL) measures.27 28
Patient’s overall satisfaction with treatment at 3 and 6 months will be evaluated with a 5-point Likert visual analogue scale (1=very satisfied to 5=very unsatisfied).29
Patient’s compliance to study drug treatment will be measured by review of the medication diary and pill count that patients will complete daily and bring to the 3-month and 6-month follow-up visits. Compliance will be considered satisfactory if at least 80% of the study drug was reportedly taken and unsatisfactory otherwise. The number of missed doses and most common reasons for non-compliance will also be documented.
SAEs will be defined using the Health Canada definition and reported as required by local regulations.30
Suspected recurrent DVT or PE will be confirmed by objective tests using criteria recommended by the ISTH.31
Success of blinding will be evaluated by asking patients, investigators and research nurses at 6 months or at the end of each patient’s participation, if earlier than 6 months, which treatment they believe they received, and the level of confidence of their responses.
Sample size and power calculations
In a recent meta-analysis of published RCTs, Kakkos and Nicolaides reported that Venixxa (Daflon), as compared with placebo, reduced the severity of leg pain, cramps and paresthesia by about 50% (relative risk (RR)=0.53, p=0.0001; RR=0.51, p=0.02; and RR=0.45, p=0.03, respectively) and leg heaviness by 65% (RR=0.35, p<0.00001).20 In Gilly et al’s and Cospite and Dominici’s RCTs, Venixxa (Daflon) was shown to reduce the intensity of symptoms and signs of CVI by at least threefold32 33 and in Chassignolle et al’s trial, by at least twofold.34 About 55% of patients on MPFF stated being very satisfied with treatment, suggesting clinically relevant improvement, as compared with only 10% of patients in the placebo group.34 Based on the above, we hypothesise that at least 50% of patients randomised to the Venixxa group will achieve the primary endpoint (ie, VS <5 or reduction in VS ≥30%). We anticipate that 20% of patients will have some PTS improvement due to a placebo effect, supported by data which suggest that on average, there could be at most a 50% mean reduction in symptoms intensity with placebo.34 A total sample size of 86 patients (accounting for a conservative 10% rate of death/loss to follow-up21 26 32–35) has 80% power to show an absolute 30% risk reduction in the primary outcome, that is, from 20% of patients improved in the placebo group to 50% of patients improved in the MPFF group, number needed to treat (NNT)=4 with a two-tailed α of 0.05.36 We will examine the impact of patient compliance to allocated treatment in secondary explanatory analyses, but not for sample size adjustment.
Statistical analyses
We will use an intention-to-treat approach for all outcomes. For the primary outcome, improvement in PTS at 6 months, a Χ2 test will be performed, stratified by centre. Tests will be two-sided with α level set at 0.05. The NNT for benefit will be calculated. In secondary analyses, we will explore the role of pre-randomisation variables (centre, duration of anticoagulation treatment and type of anticoagulation for index DVT, characteristics of index DVT: proximal vs distal; provoked vs unprovoked; bilateral vs unilateral; cancer vs non-cancer associated; and presence of contralateral vs no contralateral CVI), and post-randomisation variables (use of ECS, compliance with study drug, recurrent ipsilateral DVT during follow-up) that could influence clinical change in PTS, using logistic regression with multiple imputation to account for missing values, if necessary. In addition, adjustment for selected baseline PTS risk factors including age, sex and body mass index will be explored if there is evidence of clinically relevant differences in these variables between the two treatment groups.
For the secondary outcomes PTS severity (categorical), PTS VS (continuous), individual components (ie, symptoms, signs) of the VS, occurrence of venous leg ulcer, venous disease-specific and generic QOL, patient compliance with treatment, patients’ overall satisfaction with treatment, SAEs such as drug-related SAE, recurrent VTE and death, rates in the two intervention groups will be described, and between-group differences explored with similar procedures. For differences in mean QOL scores between groups, analysis of covariance will adjust for baseline score. Score differences on the order of 3–4 points will be considered clinically important.37 38 With 43 patients per group including a 10% dropout rate, we will have greater than 90% power to detect a 3-point difference in VEINES-QOL, assuming an SD of 9 (expected SD based on our prior work). A secondary explanatory (per-protocol) analysis will be conducted using similar procedures.
Data management
Data management will be overseen by IMS. Data will be entered online at study sites by the study nurse using a customised web-based data entry tool using validation checks at the time of data entry. Data will be reviewed and cleaned by the database coordinator on an ongoing basis by initiating and following up on queries to the sites. The cleaned database, blinded to treatment assignment, will be provided to the biostatistician for analysis, which will be carried out under the supervision of the study investigators.
Patient and public involvement
A patient partner who is a member of the Patient Partners platform of the CanVECTOR research network (www.canvector.ca) was involved in the design of this study, will participate as a member of the study steering committee and will be involved in knowledge dissemination activities after study completion.
Ethics and dissemination
The MUFFIN-PTS trial is funded by a grant from the CIHR (#332593). The study protocol was approved by Biomedical Research Ethics Committee of Centre intégré universitaire de santé et de services sociaux (CIUSSS) West-Central Montreal Research Ethics Board on 29 June 2020 (Project 2019-1656). Written informed consent will be obtained from all participating patients and consent form is provided as an online supplemental file. The MUFFIN-PTS trial is expected to begin enrolling patients in 2021. Results of the study will be disseminated via peer-reviewed publications and presentations at scientific conferences. Open access to individual patient data is not planned, but all requests for the trial’s data will be considered on an individual basis by the trial steering committee.
Supplemental material
Discussion
Given the absence of simple and effective methods to treat patients with PTS and as PTS can markedly reduce patients’ QOL, it is critical to evaluate new treatment options for patients with PTS.2 One option could be the use of venoactive drugs that are commonly used in Europe to treat CVI.
However, several considerations must be kept in mind before suggesting the use of these drugs for PTS. First, although several guidelines suggest or recommend the use of venoactive drugs to treat CVI,15 39 40 the quality of evidence suggesting their effectiveness is not high.41 In their last updated Cochrane meta-analysis focusing on venoactive drugs in patients with CVI, Martinez-Zapata et al concluded that low-to-moderate certainty evidence suggests that venoactive drugs probably reduce oedema and might, or might not, improve QOL.41 The authors underlined the need for additional high-quality RCTs focusing on clinically important outcomes. Second, the pathophysiology of PTS comprises several mechanisms, including residual venous obstruction, valvular reflux and acute DVT-related inflammation, each involved to varying degrees, depending on the individual patient’s DVT.3 The pathophysiology of PTS appears to be more complex than the pathophysiology of ‘standard’ primary chronic venous insufficiency (comprises venous reflux and some degree of inflammation). Thus, it is conceivable that treatment efficacy for a given treatment might differ between PTS and CVI, and hence results of CVI treatment studies should not be extrapolated to patients with PTS. It is therefore critical to conduct an RCT of venoactive drug specifically in patients with PTS.
We chose to test MPFF as the venoactive drug intervention for the following reasons: the literature suggests that compared with other venoactive drugs (eg, horse chestnut seed extract, ruscus extract, calcium dobesilate), MPFF—and rutosides—appear to be the most effective and safest venoactive drugs for the treatment of signs (oedema, skin changes) and symptoms related to CVI.14 42 Importantly, MPFF is the venoactive drug that has the most wide-ranging mechanisms of action for the treatment of CVI,40 including unique anti-inflammatory properties that are directly relevant to PTS pathophysiology.39 43 Furthermore, a recent Cochrane meta-analysis found no evidence that rutosides were superior to ECS or to placebo to treat PTS.44 In contrast, data on MPFF in patients with PTS are scarce but encouraging: two studies have assessed the clinical effectiveness of MPFF and both of them reported some improvement in PTS symptoms.22 23 However, these studies were small or retrospective, published in Russian language and neither was an RCT. While two small older double-blind RCTs assessed the effectiveness of MPFF (Daflon) in PTS, they did not assess clinical endpoints and rather assessed changes in venous tone, capacity and outflow.45 46 The MPFF dose that we will use (1000 mg daily) in the MUFFIN-PTS trial is the most widely used dose in published studies.20 A 1000 mg once daily formulation was shown to be equivalent to 500 mg two times per day, but this dose strength is not available in Canada.47 48
Regarding study design, given the high risk of a placebo effect in PTS studies, we opted for a double-blind design.49 Our sample size could be perceived as modest. However, based on our careful literature review, we hypothesise that there will be a 50% improvement in the primary study outcome in the MPFF arm, compared with placebo. Considering the cost of MPFF and the non-life-threatening character of PTS, we do not believe that MPFF should be used if its efficacy is only marginal. We are aware that treatment duration (6 months) might be perceived as too short to adequately assess treatment for a chronic disease. However, our 6-month treatment duration is in line with experts’ recommendations that future trials of CVI treatment should include treatment periods of at least 3 months.21 Furthermore, studies showed that the beneficial effect of MPFF may be noticed as early as 2 weeks after MPFF initiation, that the benefit increased with time and that the greatest improvements in QOL and PTS/CVI symptoms were achieved when patients were treated for at least 3 months.22 34 47 50–52 Our 6-month duration of treatment will be longer than the previous longest treatment duration (4 months) in an RCT of MPFF to treat CVI in patients without venous ulcers.20 21 If MPFF is shown to be effective to treat PTS without side effects or adverse events, longer duration of treatment will likely be needed and ideally tested.
In conclusion, the MUFFIN-PTS trial will provide definitive data on the benefits of MPFF to treat PTS. Its results should be highly relevant for patients and healthcare providers and could impact daily clinical management of patients with PTS. Indeed, providing strong evidence in support of the effectiveness and tolerability of MPFF may convince physicians to routinely prescribe MPFF to their patients with PTS, and will improve the health and QOL of patients with PTS. From the patient’s perspective, given the health burden and impaired QOL caused by PTS, a non-invasive and safe treatment to reduce the burden of PTS is desirable. From a healthcare perspective, given the high costs of treating PTS, an intervention that reduces PTS severity could be cost-effective and even cost-saving. Conversely, if no benefit of MPFF to treat is shown, many thousands of PTS patients each year around the world could be spared the cost and inconvenience of taking ineffective medications.
Ethics statements
Patient consent for publication
Acknowledgments
SK is a Tier 1 Canada Research Chair holder. All investigators are members of the CanVECTOR Network, which receives grant funding from the Canadian Institutes of Health Research (Funding Reference: CDT-142654).
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors JPG and SK wrote the protocol, designed the study and wrote the article. JA, AL-L, GLG, SSh and SSc revised and approved the protocol, critically revised and approved the article.
Funding This work is supported by a grant from the Canadian Institute of Health Research (SK foundation grant number: 332593).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.