Article Text

Abstract
Introduction Colorectal cancer (CRC) is a highly prevalent disease, wherein, ~30%–40% of patients with CRC relapse postresection. In some patients with CRC, adjuvant chemotherapy can help delay recurrence or be curative. However, current biomarkers show limited clinical utility in determining if/when chemotherapy should be administered, to provide benefit. Circulating tumour DNA (ctDNA) can measure molecular residual disease (MRD) and relapse with high specificity and sensitivity. This study protocol investigates the clinical utility of ctDNA for optimal use of adjuvant chemotherapy in patients with surgically resected CRC and to detect early disease progression in the surveillance setting.
Methods and analysis This is a multicentre prospective, observational cohort study. A total of 2000 stage I–IV patients will be enrolled in up to 200 US sites, and patients will be followed for up to 2 years with serial ctDNA analysis, timed with the standard-of-care visits. The primary endpoints are to observe the impact of bespoke ctDNA testing on adjuvant treatment decisions and to measure CRC recurrence rates while asymptomatic and without imaging correlate. The secondary endpoints are MRD clearance rate (MRD+ to MRD−) during or after adjuvant chemotherapy, percentage of patients that undergo surgery for oligometastatic recurrence, survival of MRD-negative patients treated with adjuvant chemotherapy versus no adjuvant chemotherapy (active surveillance), overall survival, examine the number of stage I CRC that have recurrent disease detected postsurgery, and patient-reported outcomes.
Ethics and dissemination This study has received ethical approval from the Advarra Institutional Review Board (IRB) protocol: Natera—20-041-NCP/3766.01, BESPOKE Study of ctDNA Guided Therapy in Colorectal Cancer (BESPOKE CRC) (Pro00041473) on 10 June 2021. Data protection and privacy regulations will be strictly observed in the capturing, forwarding, processing and storing of patients’ data. Publication of any study results will be approved by Natera in accordance with the site-specific contract.
Trial registration number NCT04264702.
- chemotherapy
- gastrointestinal tumours
- clinical trials
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
This is a large, prospective, observational study designed to examine the clinical utility of the bespoke circulating tumour DNA (ctDNA) assay, in detecting molecular residual disease (MRD) and recurrence in postresection stage I–IV patients with colorectal cancer (CRC) compared with standard of care imaging.
This protocol is designed to observe ctDNA dynamics at landmark standard of care visits, during the adjuvant and surveillance settings.
Measuring ctDNA-based MRD, postresection may aid in patient stratification by relapse risk, and aid in physician decision-making regarding adjuvant treatment decision.
This work addresses an unmet need in stage I–IV CRC, where optimal postresection treatment regimens are poorly defined, and biomarkers detecting MRD and relapse, providing real time rationale for chemotherapy initiation, escalation or de-escalation are urgently needed.
One limitation of this study is that it is strictly observational, and thus, does not dictate which type of adjuvant therapeutic intervention should be selected in the assembled cohort.
Introduction
Colorectal cancer (CRC) is the fourth most common cancer in the USA, with an estimated ~147 950 new cases emerging each year.1 CRC is also the second leading cause of cancer-related death in the USA.1 In patients with local disease, the current standard of care includes surgical resection of the primary tumour, followed by adjuvant chemotherapy in a subset of patients.2 3 For clinically high-risk (based on standard T and N criteria) patients adjuvant chemotherapy is often administered to reduce the risk of recurrence. The relative benefits of adjuvant chemotherapy have more clearly been demonstrated in stage III as compared with stage I–II CRC.3 Current guidelines recommend observation or adjuvant chemotherapy with capecitabine or 5-fluorouracil (5-FU)/leucovorin in patients with low-risk stage II disease, observation or adjuvant chemotherapy with capecitabine, 5-FU/leucovorin, FOLFOX or CAPEOX in patients with high-risk stage II disease, and an oxaliplatin-containing regimen such as FOLFOX or CAPEOX for 3–6 months in patients with stage III disease.3 The majority of stage II patients do not receive adjuvant chemotherapy, although approximately 10%–15% have residual disease following surgery.4 In addition, approximately 30% of patients with stage III disease who receive adjuvant chemotherapy also have residual disease and experience recurrence.5 Identification of patients with molecular residual disease (MRD) and treatment with additional or intensified therapy could potentially reduce their risk of recurrence. Conversely, while the majority of stage III patients receive adjuvant chemotherapy, more than 50% are cured by surgery alone and could be spared the toxicities associated with chemotherapy and/or the intensity/duration of therapy may be altered (de-intensified).6 7
Stage IV CRC is more aggressive than early-stage disease, with higher likelihood of relapse and poorer survival outcomes. Oligometastatic CRC is eligible for surgery with curative intent, but 60%–70% of patients will go on to relapse postresection.8 NCCN guidelines for stage IV disease currently recommend either adjuvant therapy or close observation during the surveillance period.3 While the benefits of adjuvant treatment have been more clearly demonstrated in the stage IV setting, the decision to pursue adjuvant therapy or observation alone is still debated.
The current standard of care highlights the need for developing better tools to facilitate physician’s decision-making in identifying and stratifying postresection patients by risk of relapse. High-risk patients would most likely benefit from adjuvant chemotherapy, while low-risk patients can safely reduce (duration/intensity) or forgo adjuvant therapy to avoid associated toxicity. In addition, sequential MRD testing (such as in patients initially found to be MRD negative, but who later become MRD positive), could help determine a time point for the initiation of postoperative therapy, potentially still with curative intent.
This hypothesis points to another significant unmet clinical need, which is early diagnosis of recurrent disease in CRC. Studies have shown that asymptomatic rather than symptomatic recurrences are more likely to be treated with curative intent and that these patients have improved overall survival after such interventions.9–11 However, despite improved outcomes for asymptomatic patients, over 60% are still diagnosed with recurrence secondary to symptoms, a scenario associated with poor clinical outcomes.12
Historically, the decision to administer adjuvant chemotherapy postsurgery and during surveillance has depended on the presence of clinical features that are considered high-risk according to the standard of care.13 This includes features observed in CT/positron emission tomography scans (bowel perforation or obstruction, etc), histological features (T4 extension, lymphovascular invasion, etc), positive staining for biomarkers from primary tumour biopsies (markers of poor differentiation, carcinoembryonic antigen (CEA) staining, mismatch repair deficiency, etc) and recently, the presence of CEA in patient serum.14 15 To date, although some of these clinical features have been associated with survival, they have been inadequate in identifying recurrence early.15 16 CEA levels are considered unreliable and may at times be elevated for a completely unrelated condition (eg, adjuvant 5-FU, cigarette smoking, gastritis, peptic ulcer disease, liver disease, chronic obstructive pulmonary diseases, inflammatory conditions and diabetes) leading to false-positive results.17–21 Overall, the poor sensitivity and specificity of CEA serum levels limits the ability to detect early-stage tumours.22 23 CT imaging is associated with radiation exposure and a real risk of secondary malignancies and has also been associated with false-positive results. In one study of 110 patients with surgically treated CRC, out of 48 events leading to suspicion of cancer recurrence, 34 (71%) were falsely positive.24
Disease recurrence is known to be associated with MRD, which arises from clinically occult micrometastases that remain in the patient during and after treatment. Blood-based evaluation of MRD by isolating circulating tumour DNA (ctDNA) from venous blood offers a non-invasive and a low-cost sampling approach that can assess disease status in real-time.25 Previous studies have demonstrated the clinical validity of ctDNA as prognostic marker that can be used to stratify patients with CRC into relapse risk groups ahead of established clinical parameters, enable early detection of recurrence and assess therapeutic response.26–30
Here, we detail a study design protocol of a prospective multicentre clinical study, to demonstrate the clinical utility of a personalised, tumour-informed ctDNA assay (bespoke, multiplex polymerase chain reaction-next generation sequencing (mPCR-NGS)) in tailoring treatment decisions in patients with stage I–III surgically resected CRC, wherein, patients with ctDNA-positivity at the MRD (postsurgical) timepoint may be considered for adjuvant chemotherapy to achieve favourable outcomes, and patients with ctDNA-negativity may reduce or forgo adjuvant chemotherapy treatment. The study will also demonstrate the clinical utility of bespoke ctDNA testing in stage IV CRC cases in the surveillance setting. These patients with increasing levels of MRD are more likely to have their disease progression detected earlier.
Methods and analysis
Overall study design
The study is a prospective, multicentre clinical study that uses a personalised ctDNA assay (Signatera bespoke mPCR-NGS), designed to track tumour-specific mutations in patients with stage I–IV CRC for MRD determination and molecular monitoring. The study started in May 2020 and is open for recruitment until October 2022. A total of 2000 subjects distributed as 500 stage I, 1000 stage II or stage III and 500 stage IV will be enrolled in up to 200 US sites. The study enrolment will begin 2 weeks prior to surgery through 6 weeks postsurgery. Patients will be followed for up to 2 years with periodic blood collection for ctDNA analysis, timed with the standard of care visits (postoperative/surgery visit, prior to initiation of systemic chemotherapy, with each cycle of chemotherapy and with each surveillance visit). Specifically, whole blood will be collected as part of routine care per the healthcare provider. Recommended follow-up draws will include week 6 (±14 days), week 12 (±4 weeks) and week 20 (±4 weeks), then every 3 months through year 2 or early withdrawal (whichever occurs first). An optional patient-reported outcomes survey will be collected at every other sample collection study visit (figure 1). Furthermore, all enrolled patients will be evaluated for any adverse events that may occur. Study inclusion/exclusion criteria are detailed in table 1.
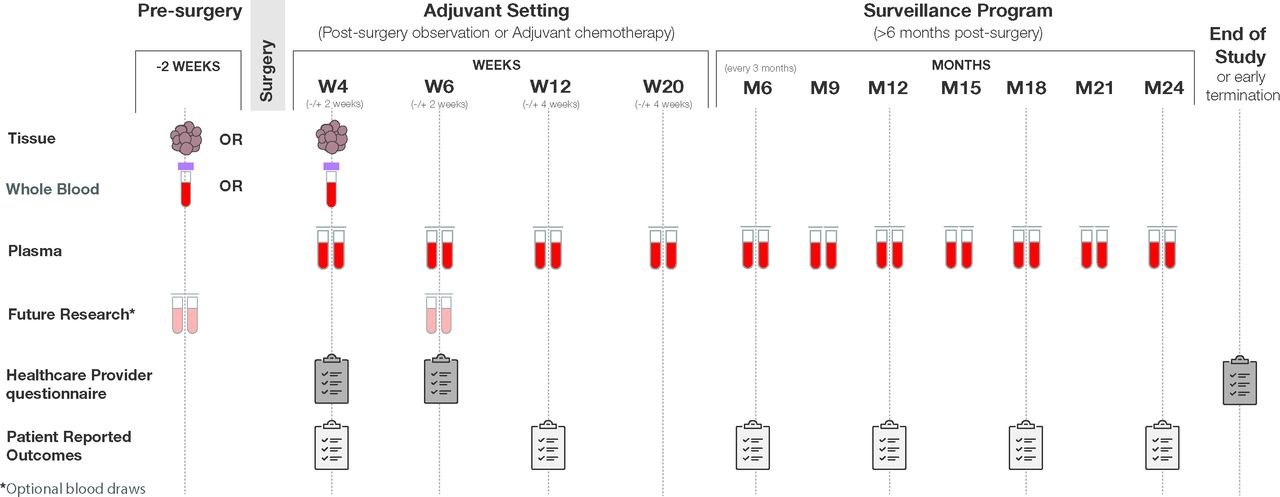
{kind=link}
BESPOKE—study design overview of the BESPOKE study design. Samples (whole blood, Formalin-Fixed Paraffin-Embedded (FFPE) tissue, plasma) will be collected, and questionnaires (healthcare provider questionnaire and patient reported outcomes), will be completed at the indicated times (weeks/months), including presurgery, during the molecular residual disease programme and during the surveillance programme.
Eligibility criteria
Control arm
Approximately 600 historical control subjects will be enrolled and will consist of matched stage I to IV CRC cases at an approximate ratio of 3:1. Each participating site will contribute retrospectively collected control patients in accordance with the site-specific study contract. Comparisons between the prospectively treated patients and the retrospective control will be conducted using inverse probability weighted data for baseline covariate adjustment. The inverse probability weights will be derived using a propensity score model that includes but is not limited to: (1) stage, (2) age and (3) gender. The retrospective controls will be patients from a similar time period such that treatment options are similar. The following criteria will be used: sex, age at the time of diagnosis (within a 10-year range) or a progression event. The patient cases would have a minimum of three evaluations per year, and a minimum of 2 years of follow-up data or had a disease progression and received treatment no more than 3 years prior to the study start date. The controls will meet all study inclusion/exclusion criteria.
Postoperative systemic therapy
Patients will be recommended to receive postoperative systemic therapy (ie, adjuvant chemotherapy) or observation at the discretion of their healthcare provider per routine practice. A list of standard adjuvant chemotherapy regimens can be found in the National Comprehensive Cancer Network (NCCN) guidelines.
Study objectives/endpoints
The primary objective/endpoint is to examine the impact of bespoke ctDNA testing on adjuvant treatment decisions, that is, the percentage of patients who have their adjuvant chemotherapy regimen increase or decrease at the first time point postsurgery, and to determine the percentage of patients who recur (ctDNA-positive at any time point, during surveillance) while asymptomatic. The secondary endpoints are to determine rate of MRD clearance (ctDNA positive at MRD time point and eventually becomes ctDNA negative during or after postoperative systemic chemotherapy), the percentage of patients undergoing surgery for oligometastatic recurrence, the survival of MRD-negative patients treated with postoperative systemic therapy versus no therapy, overall survival, and to assess patient’s reported outcomes.
Future research blood collection
An optional future research blood sample may be collected at the following time points: presurgery and/or week 6 (±2 weeks). Complete instructions for blood collection can be found in the lab manual using research collection kits provided by Natera and venipuncture will be performed using standard technique. Each collection time point will be up to 20 mL of blood and 40 mL for the entire study. All blood samples will be de-identified and include a subject ID number, sample collection date and will be recorded on the eCRF.
Data collection
As part of the protocol, demographic data, height, weight, medical and family history, and any relevant prior concomitant medication data will be recorded as shown in tables 2 and 3. At the time of enrolment, prior to receipt of ctDNA test results, the healthcare provider will complete a questionnaire regarding the disease management recommendation. Following this, patients will receive ctDNA test results, and the healthcare provider will complete a second questionnaire to record the final determination as to whether the recommendation was made for observation or postoperative systemic therapy (ie, adjuvant chemotherapy). The second questionnaire may be completed up to 20 weeks +/−4 weeks. If the recommendation was made for therapy, the specific regimen, dosing schedule and duration will also be recorded. A third questionnaire will be completed by the healthcare provider at the end of study, which will record whether the patient has cancer recurrence, has completed their 2 years of participation, or has withdrawn from the study (whichever comes first). At this point, the decision to treat, or to observe, or provide postoperative systemic therapy (ie, adjuvant chemotherapy), including the recommended regimen, dosing schedule and the duration of treatment will be at the discretion of the healthcare provider. Any unanticipated adverse events related to blood draw throughout the study will also be recorded in the eCRF. During follow-up, the following data will be recorded:
Disease status and survival.
Recurrence status (date, location and method of detection for recurrence).
Second primary cancer (diagnosis date, type of cancer and location).
Description of any additional procedures performed for the treatment of this cancer, including surgery, additional systemic therapy (ie, chemotherapy or immunotherapy) or radiation therapy.
If additional surgery performed, results of any pathology testing (a de-identified copy of the report will be provided).
If available, CEA laboratory results.
Results of any imaging studies performed since the prior visit (a de-identified copy of the report will be provided).
Diagnosis of new oncological malignancy.
Optional patient-reported outcomes may be completed at the time of enrolment and at every other sample collection study visit, or approximately every 6 months. This will include: Hospital Anxiety and Depression Scale, Fear of Recurrence, short form (FCR-4), National Comprehensive Cancer Network Functional Assessment of Cancer Therapy Colorectal Symptom Index-19 items (NCCN-FACT FCIS-19 version 2), impact of ctDNA testing results on patient’s anxiety about cancer recurrence, impact on treatment decisions and future use of ctDNA testing for monitoring cancer recurrence.
Schedule of events
Schedule of events for control arm
Data management/organisation
All data will be collected and stored in a health insurance portability and accountability act compliant fashion to maintain patient privacy. Data will be entered using an electronic data capture and will be monitored either remotely or on-site on a bi-annual basis. Prior to enrolment, signed informed consent will be received from all patients except for the control arm, wherein consent-waiver will be requested for data collection purposes. Data associated with the samples will be de-identified to maintain patient privacy. Access to the final trial dataset will be with Natera and each site will have access to their own site dataset.
Sample size and statistical considerations
The primary objective of this study is to determine the percentage of cases where the treating physician changes the postsurgical treatment regimen (increase or decrease) after obtaining the results from the bespoke ctDNA test in stage II or III subjects. Table 4 describes the assumptions considered for the percentage of patients with change in postsurgical treatment regimen and table 5 illustrates the minimum number of patients needed for this estimation with 95% confidence level by margin of error.
Assumptions for the percentage of patients with change in postsurgical treatment regimen
Minimum number of patients needed to estimate the percentage of cases with change in postsurgical treatment regimen, with a 95% confidence level, by margin of error
For this study, the percentage of cases changed in postsurgical treatment regimen will be estimated for several cohorts of patients. The following table lists each of the cohorts, the sample size requirements for each cohort and the resulting total sample size needed for the study.
Based on the sample size requirements for all CRC patient cohorts, the largest calculated sample size among the cohorts required to achieve statistical significance is 767 patients (table 6). Assuming a 20% attrition rate based on lost to follow-up, non-evaluable bespoke ctDNA results, etc), a minimum of 921 stage II–III patients with CRC will need to be enrolled in this study to meet the sample size requirements necessary to estimate the true percentage of cases with a change in postsurgical treatment regimen, under the assumptions of a ±5% margin of error and 95% confidence level.
Minimum number of patients needed in each cohort and overall, assuming a 95% confidence level and 5% margin of error for the percentage of cases changed
In addition, at least 500 stage I and 500 stage IV patients with CRC will need to be enrolled in this study based on the expected width of the 95% binomial exact CI for a binary outcome. The following expected widths for the CI based on a true rate of 50% provide the widest CI. For endpoints with 450 patients included (90% of 500), the expected CI is less than ±5%. Similarly, for numbers of patients equal to 400 (80% of 500), 300 (60% of 500) and 200 (40% of 500), the expected widths of the CIs are ±5%, ±5.8% and ±7.1%. This level of precision is considered sufficient for the evaluations of these cohorts and any observed CIs for a given subgroup may be smaller if the rate is higher or lower than 50%.
Patient and public involvement
The protocol was designed and discussed with the patient advocacy group and academic community (GI oncology). Patients and general public were not involved in the design, conduct, reporting or dissemination plans of this protocol. Patients will receive ctDNA test results from their provider, according to the current evidence-informed schedule, as part of routine practice.
Ethics and dissemination
All personnel involved in conducting the current study shall abide by the latest Declaration of Helsinki and Ethical Guidelines for Clinical Studies. Prior to enrolment, written informed consent will be obtained from all patients and compliance with all inclusion and exclusion criteria will be verified and documented. The protocol (Natera—20-041-NCP/3766.01, BESPOKE Study of ctDNA Guided Therapy in Colorectal Cancer (Pro00041473)) has been approved by the Institutional Review Board on 10 June 2021. Publication of any study results in papers, abstracts, posters or other material presented at scientific meetings or published in professional journals will be approved by Natera in accordance with the site-specific study contract.
Discussion
This study is intended to evaluate the ability of the bespoke ctDNA test for its use in CRC, including to determine MRD status postresection, to assist in the decision to administer adjuvant treatment postresection, and to monitor patients for disease recurrence and/or clearance during surveillance. Evaluation of MRD by ctDNA offers a non-invasive and a low-cost sampling approach that can be used as frequently as indicated. With a short half-life of ~2 hours, ctDNA is considered an accurate and dynamic cancer biomarker, that can be assessed in real-time to monitor tumour progression in early or advanced stage cancer, an advantage over radiographic scan.25 It has a higher sensitivity and specificity than other blood-based cancer biomarkers. Patients can be stratified by ctDNA status as an indication of risk of recurrence. Serial monitoring of ctDNA increases the likelihood of early identification of relapse, which can provide rationale for timely therapeutic intervention, before significant disease progression can occur.
The bespoke ctDNA assay (Signatera) is a personalised and a tumour-informed test, which detects MRD with high sensitivity and specificity, across all stages of CRC. The assay tracks 16 tumour-specific clonal variants in plasma based on up-front whole exome sequencing of the tumour the matched normal tissues. Unlike predesigned ctDNA static panels, a personalised, tumour-informed assay like Signatera is technically advanced as it relies on the prior knowledge of the mutational status of the patient’s tumour. Having the patient tumour tissue allows whole exome sequencing to be performed, in order to understand all of the somatic variants and select the clonal variants that are present in that patient’s tumour. By identifying and tracking clonal variants, which are expected to be present in every cancer cell from the patient, the tumour-informed approach ensures that residual disease can be detected with both a high sensitivity and high specificity, reliably detecting variants down to 0.01% variant allele frequency. The tumour-informed method also significantly reduces the false-positive rates by filtering out clonal haematopoiesis of indeterminate potential and germline-derived variants from analysis.
Various retrospective studies across cancer types, including CRC, have shown the clinical validity of bespoke ctDNA analysis to identify MRD prior to clinical or radiological relapse.31–34 A recent retrospective analysis of ctDNA in CRC showed patients (n=130) with stage I–III CRC who were ctDNA-positive at postoperative day 30 (MRD time point) were seven times more likely to relapse than ctDNA-negative patients (HR: 7.2; 95% CI 2.7 to 19.0; p<0.001). Furthermore, in the surveillance setting, ctDNA-positive patients were over 40 times more likely to recur than negative patients (HR: 43.5.0; 95% CI 9.8 to 193.5; p<0.001).32 In addition, the study also showed disease recurrence up to 16.5 months ahead of radiological imaging (average 8.7 months) with serial ctDNA analysis (sensitivity—88% and specificity 98%).32 A follow-up study by the same group enrolled 198 patients with stage I–III CRC and showed an HR of 14 (95% CI 6.1 to 30; p<0.001) in the MRD setting and an HR of 47.5 (95% CI 17.3 to 130.3; p<0.001) in the surveillance setting.35 Overall, these two studies determined that, in a multivariable analysis, ctDNA-based MRD status was the only factor significantly associated with relapse-free survival, after adjusting for all other standard clinicopathological factors. A clinical experience study with ctDNA testing showed assessment of MRD rates across patients with early and advanced stage CRC, wherein postsurgical MRD rate was observed to be significantly higher in patients with high-risk stage II, T4N0 (28.6%; 4/14) compared with low-risk stage II T3N0 (5.6%; 3/53) and a similar trend was observed with postsurgical stage III, high-risk T4, N1-2, T-Any, N2 MRD rate (39.4%; 15/38), compared with stage III, low-risk T1-3N1 (9.3%; 3/32). This further emphasises the clinical utility of ctDNA (personalised, tumour-informed) analysis for accurately detecting ctDNA especially at the postsurgical MRD timepoint.36 Our most recent study stage IV patients with CRC found that postresection ctDNA status was predictive of disease progression, with a sensitivity of 72% and a positive predictive value of 96.7%. MRD-positivity was associated with a marked reduction in disease-free survival (DFS) (HR: 5.8; 95% CI 3.5 to 9.7; p<0.001) and overall survival (OS) (HR: 16.0; 95% CI 3.9 to 68.0, p<0.001). Additionally, in a subset analysis, ctDNA-negative patients not treated with systemic therapy showed an OS of 100%).37 This demonstrates the clinical opportunity to safely avoid adjuvant therapy in MRD-negative patients, while continuing to monitor these patients over time.
Incorporation of ctDNA testing in clinical trials provides an opportunity of being used as a new surrogate endpoint for treatment efficacy and disease status. Additionally, if used in conjunction with surveillance tools, a positive ctDNA test can help resolve ambiguous findings and may justify initiation of chemotherapy. This is likely when the recurrent tumour is still in its early stages and is too small to be detected by imaging. Conversely, a ctDNA-negative result, may reduce the patient’s exposure to unnecessary chemotherapy and avoid associated toxic side effects and comorbidities. Of note, a recently published clinical trial protocol report by Taniguchi H, et al demonstrate the use of tumour-informed ctDNA testing in a prospective, multi-centre randomised trial (CIRCULATE—Japan) to assess MRD status in postoperative patients with CRC (stage II–IV) and stratify them into therapeutic escalation and de-escalation arms.38 The study showed a significantly inferior DFS for MRD-positive both at single postsurgical 4 weeks time point and (HR: 19.5, p<0.001) and at any time point (4-week, 12-week or 24-week) (HR: 46.8, p<0.001).39 Thus, ctDNA testing combined with these insights can yield even greater improvements in treatment decisions. ctDNA testing is also being used in a growing number of other ongoing CRC clinical trials for evaluating efficacy of novel treatment strategies, enrichment of high-risk patients in trials or as a surrogate endpoint (NCT03748680, NCT04068103, ACTRN12615000381583, NCT04259944, ACTRN12617001566325, NCT03803553, NCT03844620, NCT03436563).
With respect to patient outcomes, the current 5-year survival rate for CRC is approximately 64%.40 Poor outcomes associated with CRC are in part due to the subset of patients that present with advanced CRC and show treatment resistance, as well as the limitation of existing standard of care to detect and treat recurrence early. Although it is not the main goal of the present study, assessment of the impact of bespoke ctDNA testing on patient survival is of interest going forward. It may be useful to follow up with patients beyond the conclusion of this 2-year study, to accurately assess the impact of the bespoke ctDNA test, and the resulting treatment decisions made, on CRC survival outcomes.
This study is limited by its purely observational design, which does not direct therapeutic intervention or which type of therapy to select. However, this study will measure the impact of ctDNA testing on physician decision-making, including treatment escalation or de-escalation. Furthermore, while it has been suggested that ctDNA testing may result in patients receiving less chemotherapy than is needed for optimal treatment, our protocol is designed to promote treatment optimisation. For patients who are ctDNA MRD+, we recommend that the treating physician administer/escalate chemotherapy. For those who are ctDNA MRD−, it is important to note that our protocol is designed in a way that these patients would have already started adjuvant chemotherapy, as prescribed by their treating physician, at the time of initial ctDNA analysis. De-escalation of chemotherapy, if any, will be pursued at the discretion of the treating physician, and ctDNA status be routinely monitored, to verify disease clearance, over time.
Overall, this study will set the benchmark for providing ctDNA MRD-positive rates in colorectal cancer patients postresection, as well as ctDNA kinetics in response to adjuvant chemotherapy. Patients with rectal cancer who only received traditional chemoradiation but not induction chemotherapy or total neoadjuvant therapy are eligible to enrol in this study. Their participation will help elucidate the benefit of further adjuvant chemotherapy in this subset of patients.
Ethics statements
Patient consent for publication
Acknowledgments
Authors would like to acknowledge the clinical project management and data management support provided by Worldwide Clinical Trials.
References
Footnotes
Twitter @pashtoonkasi
Contributors Study coordinator: SS, JG. Site Identification: NH, SKr. Design and writing of the protocol: AA, SS, SE, JW, AR, SKo, PMK, AG. Data collection: MMu. Data analysis: JW. Data interpretation: N/A. Writing of the manuscript: AKM, MMa. Statistical setting of the study design and data analysis: JW. All authors reviewed and approved the final manuscript: PMK, SS, JG, MMu, SE, JW, NH, SKr, AKM. MMa, AR, SM, AG, SKo, PB, AA.
Funding This study was supported by Natera, Inc. This study received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests PMK: consultancy/advisory board: Taiho Oncology, Ipsen, Natera, Foundation Medicine; Research/Trial Support (to institution): BMS, Celgene, AstraZeneca, BTG, Advanced Accelerator Applications, Array Biopharma. AG: Research funding from Array BioPharma, Bayer, Boston Biomedical, Daiichi Sankyo, Eisai, Genentech/Roche, Lilly, Pfizer. SKo: research funding from Amgen, Array BioPharma, Biocartis, EMD Serono, Genentech/Roche, Guardant Health, Lilly, MedImmune, Novartis, Sanofi. All other authors are employees of Natera, Inc. with stock/options to own stock on the company. This study is being sponsored by Natera, Inc.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.