Article Text

Abstract
Introduction The Cognitive Health in Ageing Register: Investigational, Observational and Trial Studies in Dementia Research (CHARIOT): Prospective Readiness cOhort (PRO) SubStudy (CPSS), sponsored by Janssen Pharmaceutical Research & Development LLC, is an Alzheimer’s disease (AD) biomarker enriched observational study that began 3 July 2015 CPSS aims to identify and validate determinants of AD, alongside cognitive, functional and biological changes in older adults with or without detectable evidence of AD pathology at baseline.
Methods and analysis CPSS is a dual-site longitudinal cohort (3.5 years) assessed quarterly. Cognitively normal participants (60–85 years) were recruited across Greater London and Edinburgh. Participants are classified as high, medium (amnestic or non-amnestic) or low risk for developing mild cognitive impairment–Alzheimer’s disease based on their Repeatable Battery for the Assessment of Neuropsychological Status performance at screening. Additional AD-related assessments include: a novel cognitive composite, the Global Preclinical Alzheimer’s Cognitive Composite, brain MRI and positron emission tomography and cerebrospinal fluid analysis. Lifestyle, other cognitive and functional data, as well as biosamples (blood, urine, and saliva) are collected. Primarily, study analyses will evaluate longitudinal change in cognitive and functional outcomes. Annual interim analyses for descriptive data occur throughout the course of the study, although inferential statistics are conducted as required.
Ethics and dissemination CPSS received ethical approvals from the London—Central Research Ethics Committee (15/LO/0711) and the Administration of Radioactive Substances Advisory Committee (RPC 630/3764/33110) The study is at the forefront of global AD prevention efforts, with frequent and robust sampling of the well-characterised cohort, allowing for detection of incipient pathophysiological, cognitive and functional changes that could inform therapeutic strategies to prevent and/or delay cognitive impairment and dementia. Dissemination of results will target the scientific community, research participants, volunteer community, public, industry, regulatory authorities and policymakers. On study completion, and following a predetermined embargo period, CPSS data are planned to be made accessible for analysis to facilitate further research into the determinants of AD pathology, onset of symptomatology and progression.
Trial registration number The CHARIOT:PRO SubStudy is registered with clinicaltrials.gov (NCT02114372). Notices of protocol modifications will be made available through this trial registry.
- epidemiology
- dementia
- preventive medicine
- old age psychiatry
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
Prospectively designed, high-powered longitudinal cohort of cognitively healthy (at baseline) elders across the Alzheimer’s pathological continuum followed up at high throughput using biological, psychosocial, cognitive, behavioural and lifestyle measures.
Study adopts a unique cognition-based classification method for designating risk of mild cognitive impairment–Alzheimer’s disease development from baseline.
Given the low amyloid positivity rate and the requirement of an equal number of CPSS participants above and below threshold, a high number of participants (78.6%) were excluded from the longitudinal CPSS study.
The conduct of the study at only two sites does not fit the model of a typical, multisite international clinical trial.
Introduction
Background and rationale
The last few decades have witnessed unparalleled growth in aged populations. Hence, the global incidence and prevalence of Alzheimer’s disease (AD), the most common form of late-onset dementia, continue to increase exponentially, with numbers expected to exceed 150 million global cases by 2050.1 The paucity of any viable therapy for dementia prevention and/or disease modification necessitates a rethink of the conventional approach towards preventative research. Indeed, the AD field will benefit from concerted efforts for preventative strategies combining biomarker discovery studies with detailed validation of clinical characteristics as well as longitudinal explorations of associated pathologies and symptoms.
The asymptomatic stage of AD is characterised by biomarker evidence of amyloid-β (Aβ) deposition, as measured by either low cerebrospinal fluid (CSF) Aβ42 peptide concentrations or elevated tracer uptake on Aβ positron emission tomography (PET) scans.2 Multiple studies have now reported that higher Aβ burden in cognitively normal (CN) individuals is associated with measurably poorer performance in neuropsychological tests.3 The accumulating longitudinal data also strongly suggest that evidence of abnormal levels of Aβ deposition in CN individuals increases the risk for cognitive decline and progression to mild cognitive impairment (MCI) and AD dementia.3 The current consensus among members of the Alzheimer’s scientific community is that these CN individuals with detectable pathogenic Aβ represent an early stage on the AD continuum.2 4 5 Indeed, a meta-analysis of 55 studies suggested that approximately 20% to 35% of study participants aged over 60 years without dementia symptoms are likely to have above-threshold pathogenic Aβ pathology detected by PET,6 with numbers increasing to 90% by age 85.7
Rate of cognitive decline in CN individuals with or without evidence of abnormal Aβ deposition can be measured using sensitive cognitive composite instruments. These measures focus on the cognitive domains affected earliest in AD, namely episodic memory and executive function, with decline noted as early as 7–10 years prior to the diagnosis of MCI or AD dementia.8–10 Yet, gaps remain in our understanding of the exact predictors of AD pathological onset, accumulation and resultant development of clinical symptoms. There is a need to identify individuals at varying levels of risk for AD, prior to development of AD dementia. Such information would be useful to improve our understanding of the natural history of AD progression and identify opportunities for intervention.
The CHARIOT:PRO programme seeks to address such gaps via detailed explorations of the determinants of AD-related biological, clinical and cognitive changes. The previously reported main study of 987 participants at ICL, conducted from 2013 to 2016 (following early termination by the study sponsor)11 was further adapted into a large prospective observational trial—The CHARIOT: PRO SubStudy (CPSS) aimed at enhancing the scientific robustness of the main study objectives with the addition of imaging and other AD-related assessment tools.
Here, we describe the protocol (from Amendment version 15, dated 15 Aug 2018) of this biomarker enriched CPSS featuring neuropsychological, functional, lifestyle, imaging and other biomarker assessments and the schedule for their collection. We provide an outline of the study design and a summary of the recruitment and screening process leading to the fully enrolled cohort of 519 cognitively unimpaired adults.
Objectives of the CPSS
CPSS is a prospective dual-centre, UK cohort study that at its core aims to characterise deeply the clinicobiological attributes of the non-symptomatic AD stage in individuals at differing levels of risk for development of MCI and AD dementia, based on cognitive test scores at screening. CPSS participants thus could form a readiness cohort to be recruited onto future AD dementia prevention trials.
Specifically, using data from participants with evidence of detectable Aβ pathology versus those with below-threshold levels, the study will:
Investigate the longitudinal change of the global and composite measures of the newly-developed Global Preclinical Alzheimer’s Cognitive Composite (G-PACC) in comparison to the Repeatable Battery for the Assessment of Neuropsychological Status (RBANS)12 and other study neuropsychological assessments, as well as psychometrically evaluate the test batteries.
Determine precise baseline predictors of longitudinal AD-related cognitive and functional decline, and clinical progression to improve future screening of participants most likely to develop MCI-AD/AD dementia.
Methods
Population
The CPSS participants are adults aged 60–85 years old (inclusive), residing in Greater London, South West England, Edinburgh and surrounding districts. Those included had documented evidence of Aβ pathology (Aβ positives: above-threshold brain Aβ deposition on PET or below-threshold CSF Aβ42 concentration), or evidence of below-threshold Aβ pathology (Aβ negatives: below-threshold brain Aβ deposition on PET or above-threshold CSF Aβ42 concentration), and a baseline global Clinical Dementia Rating (CDR) score=0. CPSS participants were classified at screening as high, medium-amnestic or non-amnestic, or low risk for developing MCI due to AD (MCI-AD), based on cognitive test performance as described previously.11
Study design
The CPSS is a UK prospective observational study taking place across two sites (ICL and EDI). The study is planned to follow approximately 250 CN participants who are Aβ positive and approximately 250 Aβ negative CN control participants for up to three and a half years. Evidence of Aβ pathology was assessed via CSF Aβ42 except where lumbar puncture (LP) was medically contraindicated or refused by participants, in which case Aβ PET was permitted as an alternative method of determination of Aβ status. CSF samples were tested with the Meso Scale Discovery (MSD) triplex (Aβ38/40/42). A binary classification for Aβ load was applied using a cut-off value for CSF Aβ42 ≤ 600 ng/L. The cut-off for brain Aβ PET via standardised uptake value ratio (SUVR) was based on three independent F18-radiolabeled amyloid tracers—florbetapir, flutemetamol and florbetaben. A specific SUVR threshold (ie, a cut-point) was used for each of the three radiotracers (Amyvid: 1.14 with whole-cerebellum as a reference region, Neuraceq: 1.20 with cerebellar grey matter, Vizamyl: 1.23 with whole cerebellum). Scans were reported as amyloid positive if the composite cortical SUVR value was above the defined tracer-specific threshold, and negative if less than or equal to the threshold value.
All study investigators, sponsor team and participants are blinded as to Aβ status information, with the exception of an unblinded team member for verification of imaging and CSF Aβ information. Blinding was put in place to avoid bias for conducting, monitoring and interpreting results from the clinical assessments (except for research analysis purposes). The same double blind is maintained for apolipoprotein E (APOE) genotype, in view of allele-specific positive correlation with Aβ load.13–15 Aβ status and APOE genotype results were not disclosed to participants as the clinical value (ie, diagnostic or predictive) of such a disclosure in a CN population is still unestablished. If clinical value is established from this or other studies, then amyloid and APOE genotype will be disclosed to participants, at the end of the study.
Numbers of Aβ negative participants who passed screening assessments were deliberately controlled to ensure equivalency with number of eligible Aβ positive participants. There was no deliberate effort to balance the groups by age or gender.
Study schedule
Participant recruitment
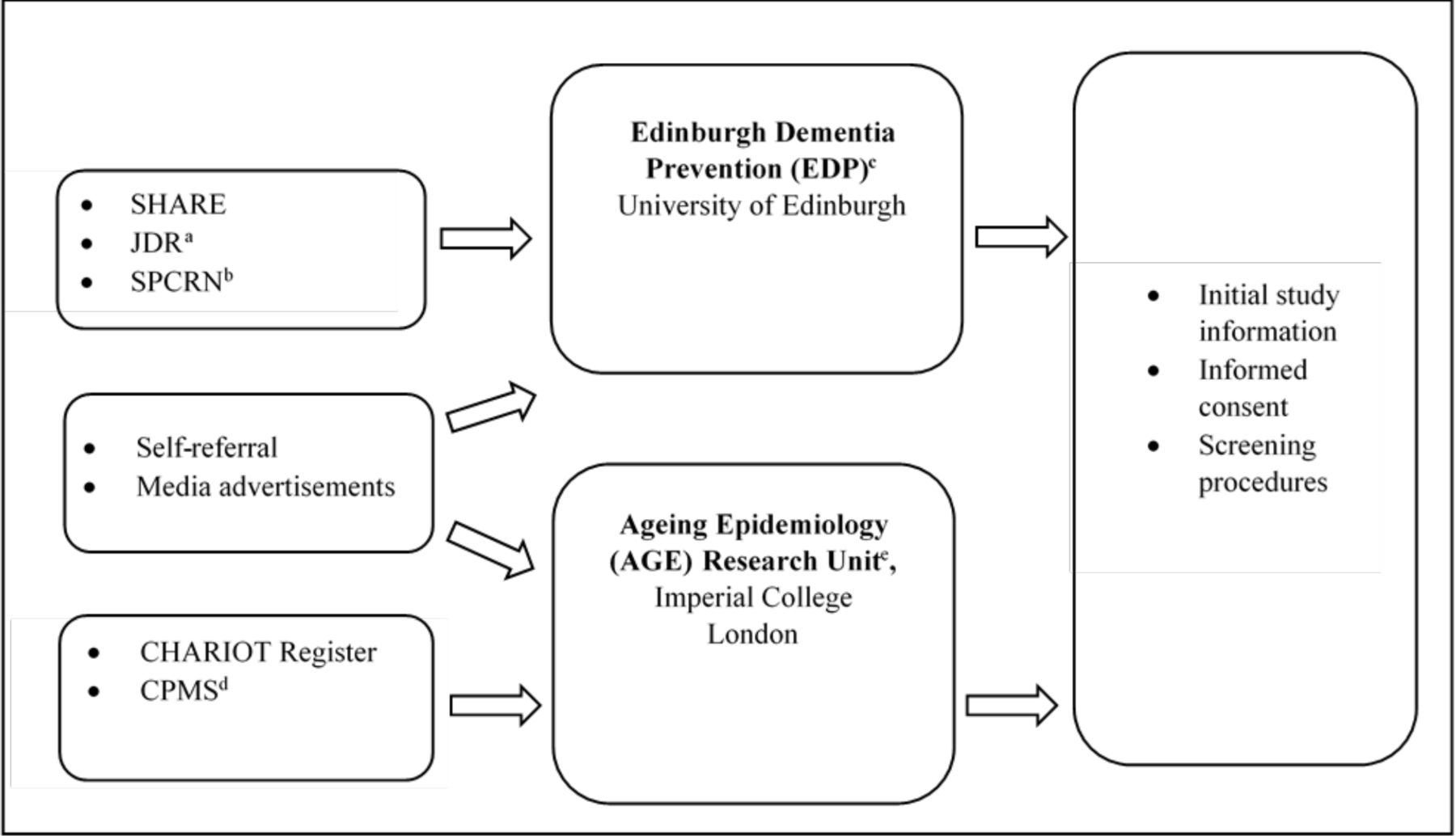
At the ICL site, participants were recruited primarily from the CHARIOT Register, a well-established dementia prevention and prediction register of older adults without dementia who have provided consent to be contacted for relevant ageing research.16 17 Some participants transitioned directly to CPSS from the main study, though most of these individuals had previously been recruited also from the CHARIOT Register. Additional methods of recruitment at the site, with very limited numbers of enrolled participants, included self-referrals and response from media advertisements. At the Edinburgh site, participants were recruited via SHARE (https://www.registerforshare.org), Join Dementia Research (JDR, https://www.joindementiaresearch.nihr.ac.uk/) and the Scottish Primary Care Research Network (SPCRN http://www.nhsresearchscotland.org.uk/research-areas/primary-care/about-the-network) (see figure 1 for the participant recruitment pathway). Recruitment efforts resulted in 1914 individuals screened at ICL to enrol 409 participants, and 537 screened at Edinburgh to enrol 110. Screened participants were not selected based on race/ethnicity or gender, resulting in a predominance of participants of European ancestry (>95%) and a slight majority of women.
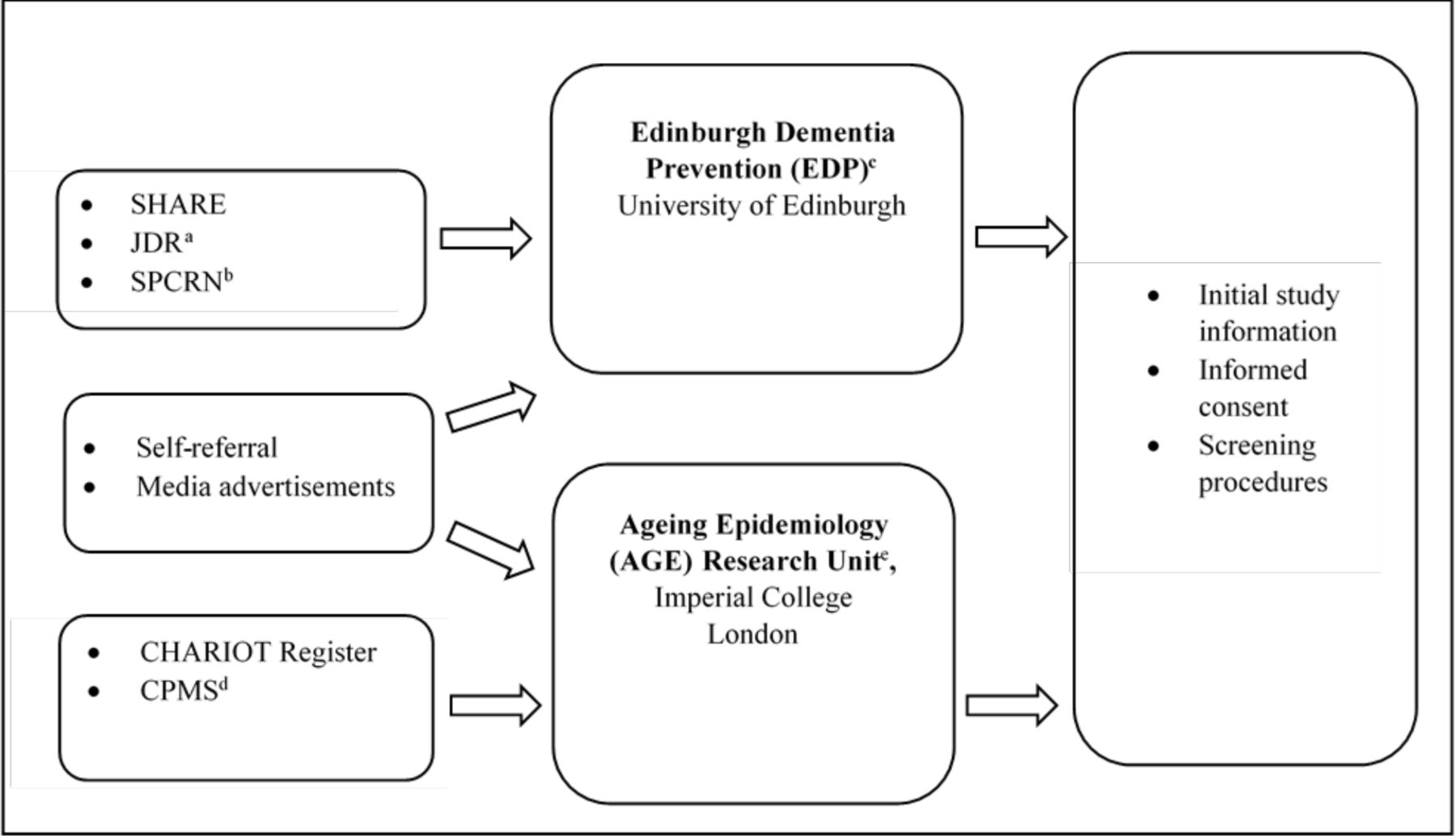
The Cognitive Health in Ageing Register: Investigational, Observational and Trial Studies in Dementia Research (CHARIOT): Prospective Readiness cOhort (PRO) Substudy Recruitment Pathway. aJoin Dementia Research. bScottish Primary Care Research Network. cFormerly Centre for Dementia Prevention. dCHARIOT:PRO Main Study. eFormerly Neuroepidemiology and Ageing (NEA) Research Unit.
Selection of study participants: summary of eligibility criteria
The major exclusion criteria for CPSS include known familial autosomal dominant AD, diagnosis of AD dementia, MCI, or any other degenerative brain disorder that is associated with dementia at screening. Evidence of brain disease or other conditions leading to dementia, other than AD-related structural pathologies were assessed centrally by blinded neuroradiologists via MRI during screening. Additionally, use of AD pharmacological therapies, and evidence of psychiatric/cognitive disorders/other abnormalities such as low vitamin B12 (specifically those with abnormal homocysteine and methylmalonic acid), and linked to cognitive deficits are exclusionary. Further, history of first-degree family member with diagnosed clinical AD was required for participants aged 60–65 years. This measure was put in place to enrich the cohort for cerebral Aβ positivity given typically lower prevalence in asymptomatic young elders, that is, below 70 years of age,18 thereby effectively minimising screen failure rates. Following participants’ consent, self-reported medical and medication history was confirmed from full history provided by participants’ general practitioner (GP). On receipt of any medical information, current medical conditions and medical history was updated on source documents and subsequently on electronic data, including medication, past and planned procedures. Medical summaries from GPs were used to ascertain self-reported histories.
During screening, participants whose cognitive performance on any RBANS Index fell more than 1.5 SD below the (age and education adjusted) population mean (based on normative sample from19) were referred to an adjudication panel. This panel, comprising neurologists, psychiatrists and neuropsychologists, considered whether the low performance was likely to be attributable to undiagnosed cognitive impairment and, if so, excluded the participant from the study. These participants were contacted directly by the study team to inform them of their exclusion. At that time, where any concerns were noted regarding their performance, the option to notify their GP with information about the study and their exclusion was offered.
Screening schedule
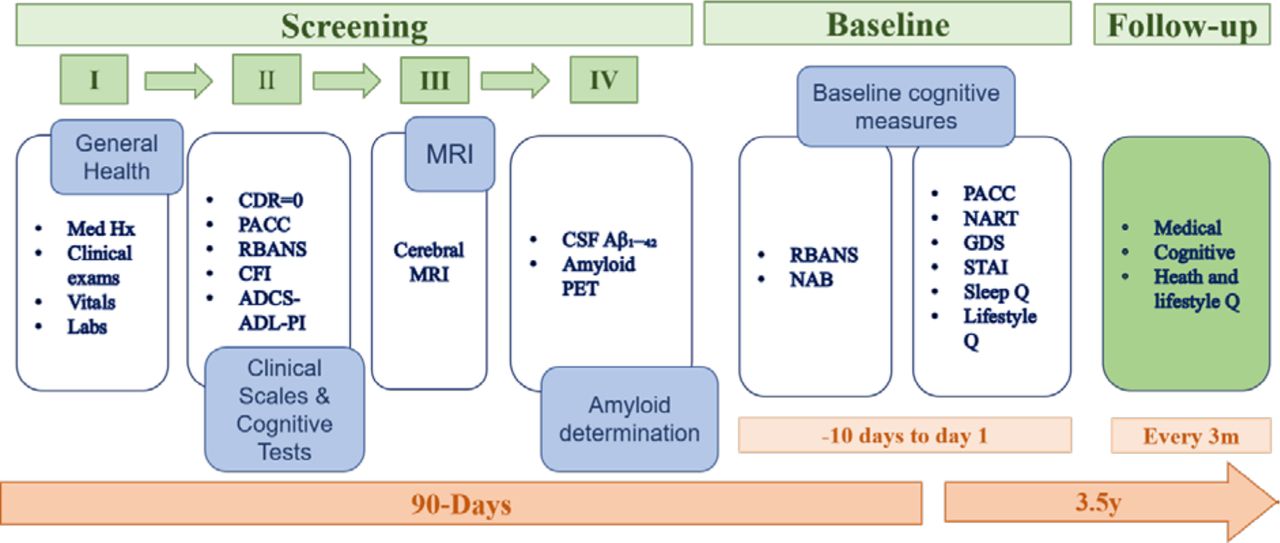
The screening was usually completed in four separate visits within a 90-day window. On certain occasions, this timeline was extended up to 180 days to allow for treatment of transient conditions, laboratory retesting and scheduling of other screening assessments. This allowed time for results to be received and evaluated against study eligibility criteria. Any clinically significant findings were passed on for follow-up to the participant’s GP. Participants who were determined to have an active unstable illness, as defined by the inclusion/exclusion criteria, were excluded. Screening involved collection of demographic data which included age, ethnicity, education and occupational status. During screening, potential participants completed cognitive tests, the G-PACC and RBANS, and CDR including the study partner interview. A clinical evaluation (pulse, blood pressure, weight, head, waist and hip circumference, temperature (tympanic), physical and neurological examination) and clinical lab assessments were carried out to determine general health status. Participants not excluded at this stage then underwent a brain MRI. If MRI did not reveal exclusionary abnormalities (see box 1), it was followed by an Aβ assessment based on CSF analysis or brain PET scan. After the Aβ determination, baseline assessments were undertaken at two consecutive visits where the RBANS (form A), G-PACC (form A) as well as the Neuropsychological Assessment Battery (NAB) Memory and Executive Function modules (form 1) and the National Adult Reading Test (NART) were administered alongside self-reported study questionnaires. Biosamples were further collected for biomarker assessments (see figure 2 for schematic depiction of screening and baseline assessments).
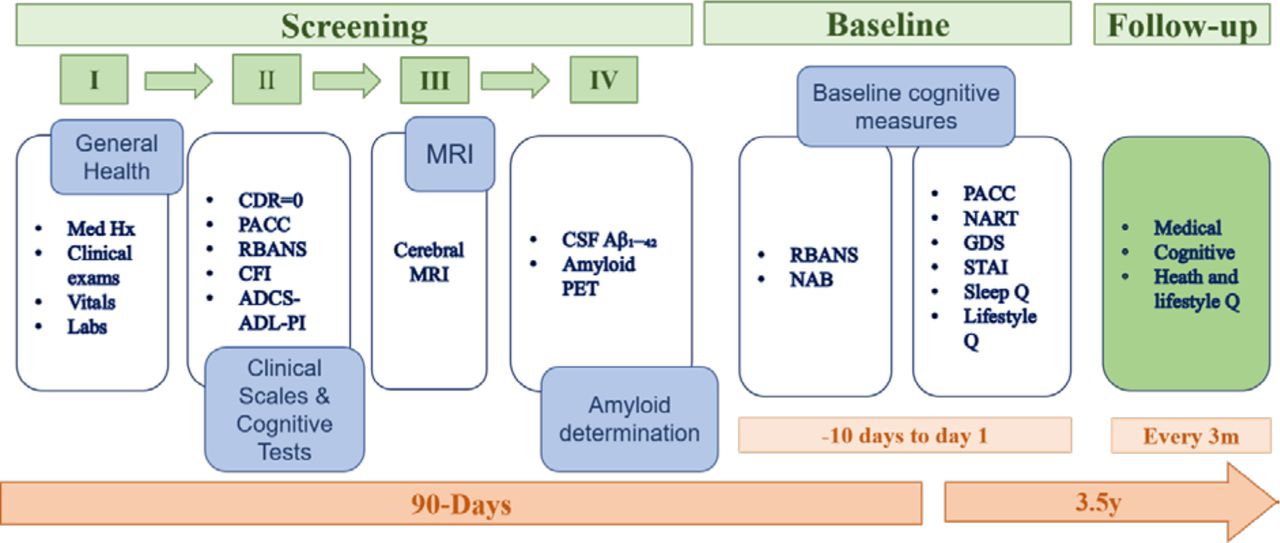
Screening and baseline assessment schedule. CDR, Clinical Dementia Rating; CSF, cerebrospinal fluid; PACC, Preclinical Alzheimer Cognitive Composite; PET, positron emission tomography; RBANS, Repeatable Battery for the Assessment of Neuropsychological Status; CFI, Cognitive Function Index; ADCS-ADL-PI, ADCS-Activities of Daily Living—Prevention Instrument; NAB, Neuropsychological Assessment Battery; NART, National Adult Reading Test; GDS, Geriatric Depression Scale; STAI, State Trait Anxiety Inventory.
Cognitive Health in Ageing Register: Investigational, Observational and Trial Studies in Dementia Research: Prospective Readiness cOhort study exclusionary findings post screening MRI
Oedema including amyloid-related imaging abnormalities (ARIA-E)
Hydrocephalus
>25% age related white matter disease
Frontal or temporal atrophy not typical of AD
History or evidence of a single prior haemorrhage >1 cm3
Multiple lacunar infarcts (two or more) or
Single prior infarct >1 cm3
Cerebral contusion, encephalomalacia
Aneurysms, vascular malformations
Subdural haematoma
Space-occupying lesions (eg, abscess or brain tumours such as meningioma >1 cm)
MRI features atypical of AD dementia.
*Evidence of brain oedema (eg, ARIA-E, vasogenic oedema, hemosiderin deposits (HD)≥10 mm in size or HD <10 mm in size but >10 in number) will be reviewed by the Sponsor’s Medical Monitor to address plans for clinical evaluation and follow-up as well as for potential inclusion/exclusion in the study.
Postscreening schedule
Following the baseline assessment, CPSS participants were randomised in a balanced 1:1:1 ratio, stratified by Aβ status and level of performance on the screening RBANS, to one of three supplemental neuropsychological tests namely: CogState Brief Battery,20 Cognitive Drug Research Assessment System, and either Delis-Kaplan Executive Function System (DKEFS, 21, ICL only) or COGNITO (22, EDI only). Participants who enrolled in the Substudy from the Main Study retained their previous Main Study-assigned randomised group. Participants are expected to attend study visits every quarter and will be followed up for a period of up to 3.5 years. Due to the COVID-19 restrictions that were implemented in March 2020 in the UK, the CPSS was transitioned to virtual visits to allow continued longitudinal assessments. For further details on our strategy for operationalising this activity, see Ref 21. As part of the general visits, we collect detailed information on all medical, especially COVID-19-related incidents including more recently information on COVID-19 vaccinations. These data are designated COVID-19-related within our database for easy identification of such cases.
Study outcomes and assessments
Primary neurocognitive outcomes
The primary outcomes of the CPSS are performance in two neurocognitive measures, the novel G-PACC and the RBANS.
The G-PACC: is a retrospectively and theoretically derived and validated measure, weighted towards episodic memory but including a timed executive function test and a global cognitive screening test.22 For this study, the four PACC components include: the Free and Cued Selective Reminding Test—Immediate Recall (FCSRT-IR),23 the Delayed Paragraph Recall score on a single administration of the Logical Memory story from the WMS—Revised,24 the WAIS-IV Coding subtest24 and the MMSE.25 Each component score is transformed into z-scores. These z-scores are summed to form the composite. The battery takes about 25 min to administer. Alongside screening and baseline time points, alternating forms of the G-PACC are administered at the following time points: months 6, 12, 18, 24, 30, 36, and 42.
The RBANS: is a 25 min composite battery with 12 subtests that measure 5 cognitive domain indices: Attention, composed of Digit Span and Coding, Language, with Picture Naming and Semantic Fluency subtests, Visuospatial Construction including Figure Copy and Line Orientation subtests, Immediate Memory comprising List Learning and Story Memory subtests, and Delayed Memory composed of List Recall, List Recognition, Story Recall and Figure Recall subtests. The sum of these 5 Index scores is converted to a Total Scale value via a mapping table. The Total Scale is a norm-based t-score based on a distribution with a mean of 100 and SD of 15. The RBANS is administered face-to-face, has 3 alternate forms, is available in over 30 languages, and has been used in multinational clinical trials including AD trials. Alternating forms of the RBANS are also administered at the following timepoints: months 3, 9, 15, 21, 27, 33, and 39. During screening, participants’ RBANS scores were used to delineate risk (low, medium, high) for developing MCI-AD, as described in the Main study.11
Secondary cognitive outcomes and functional outcomes are described in table 1. More detailed description of these measures are provided in the CHARIOT PRO Main Study Protocol.11
Cognitive Health in Ageing Register: Investigational, Observational and Trial Studies in Dementia Research (CHARIOT): Prospective Readiness cOhort (PRO) study Cognitive and Functional outcomes
Neuroimaging outcomes
Safety and volumetric scans (3DT1, FLAIR, T2*, PD/T2, T1 and DWI)
All potential CPSS participants underwent brain MRI at screening to assess eligibility, based on a central radiologist’s interpretation of the MRI scan under the supervision of Bioclinica. Borderline findings were reviewed by the Medical Monitor prior to determining participant eligibility. Image acquisition was performed at multiple sites based on a standardised MRI protocol. General Electric Signa HDxt 1.5T and Siemens TrioTim, Verio, Skyra and Prisma 3T scanners were used to acquire a volumetric 3D T1 weighted series in a sagittal plane, using 1.2 mm thick slices and a 192×192 acquisition matrix over a square FOV of 240 mm. Contrast parameters were field strength and manufacturer dependent (Siemens MP-RAGE and GE IR-Prep Fast SPGR). The standardised MRI protocol also included 2D axial FLAIR, T2* gradient echo, dual-echo proton-density and T2-weighted turbo/fast spin echo, T1-weighted turbo/fast spin echo and diffusion-weighted imaging. Proper implementation of the MRI protocol on each participating scanner was verified prior to first subject scan by use of American College of Radiology (ACR) phantom scans.
Exploratory scans (task-free BOLD functional MRI (tf-fMRI) and high-resolution coronal T2 sequences)
At the ICL site, the first 800 subjects who were eligible for MRI underwent a dual-echo GRE field map and task-free functional MRI time series. For the remainder of the subjects, a high-resolution two-dimensional (2D) coronal T2-weighted sequence was acquired, in order to visualise hippocampal subfields.
Amyloid-β positron emission tomography
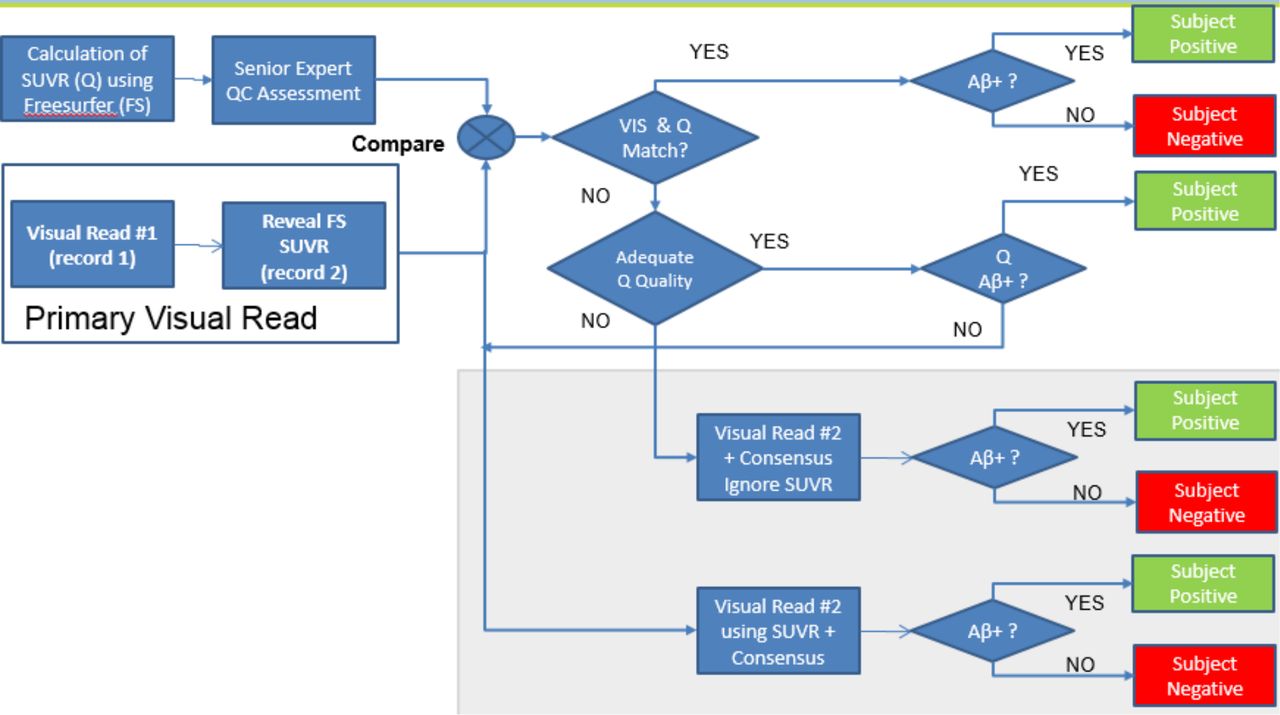
At final stage of screening, evidence of Aβ pathology in potential CPSS participants was assessed by a brain PET scan. All images derived were evaluated centrally at Bioclinica for Aβ status assessment. The assessments were performed by neuroradiologists trained in the assessment of Aβ PET scans using F18-radiolabeled amyloid tracers (Amyvid, Vizamyl and Neuraceq) for amyloid status according to the reading process developed by radiotracer vendors. The PET scan was evaluated at baseline to determine each patient’s Aβ status as positive or negative and therefore inclusion or exclusion into the trial. PET exams were acquired using a uniform scanning protocol that minimises and accounts for between-site differences in PET systems, as characterised with a Hoffman phantom exam. All exams were acquired in 3D mode and employed correction for attenuation, scatter and random coincidence. Semiquantitative SUVR assessment was performed prior to the visual read. SUVR calculations leveraged a FreeSurfer-based native-space MRI segmentation method. The Aβ status assessment was a hybrid visual and quantitative approach (see figure 3). A visual review was performed by a single reader, followed by positivity assignment based on SUVR cutpoint. In case of discrepancies between visual and SUVR results, a second reader was asked to participate in a final decision on amyloid status, as part of a consensus review. The second reader was given both the initial visual read and the SUVR measurement and convened with the first reader to arrive at a consensus assessment.

{kind=link}
{kind=link}
{kind=link}
Positron emission tomography (PET) amyloid-β (Aβ) status reading workflow. SUVR, Standardised Uptake Value Ratio; Q, quantification of SUVR; FS, parcellation of cerebral structures based on Freesurfer imaging pipeline; QC, quality control assessment; VIS, result of visual assessment of amyloid PET; Aβ+, assessed as Aβ positive based on visual and/or quantitative (SUVR) analysis of amyloid PET.
Fluid biomarkers
Blood, saliva and urine samples for clinical assessments and future biomarker discovery studies
At ICL and EDI, blood and urine samples were collected at screening to assess general health status. These included: haematology and differential panel, lipid panel, chemistry panel, electrolyte panel, coagulation group, C reactive protein, TSH, folate, vitamin B12 and urine macropanel (with urine microscopy if abnormal macro panel).
At ICL, serum, plasma, buffy coat, whole blood, urine and saliva samples are processed and stored at baseline and annually thereafter for future biomarker exploration. Samples for biobanking are collected between 9:00 and 11.30 and following an overnight fast; and are stored at the ICL purpose built −80°C biobank for future analyses. All samples are processed within 2 hours of collection, as per guidelines on biomarker preprocessing.26 Planned analyses include untargeted metabolite and proteome profiling, to generate novel targets for future hypothesis testing and biomarker discovery studies.
CSF biomarkers
For those participants not receiving Aβ PET, CSF samples were collected during screening and analyses for AD-related markers including beta-amyloid, total tau and phosphorylated tau. The Aβ data were used for determination of enrolment eligibility, and in addition to the tau data, will be useful for disease modelling and staging of preclinical AD per NIA-AA criteria.2 At ICL, additional aliquots of CSF samples are stored in the −80°C biobank for future analyses, which may include the exploration of putative biomarkers of AD pathophysiology as they arise in the literature.
Genetic outcomes
Whole blood is collected in EDTA tubes for extraction of genomic DNA (gDNA) using standard methods. gDNA were thus isolated via commercially available kit following manufacturer instructions (QIAgen QIAsymphony DSP DNA Mini Kits or Promega Maxwell RSC Whole Blood DNA Kit). Both kits facilitate automated magnetic bead-based extractions that successfully extract DNA from human whole blood samples with good quantitation and purity assessments. The QPS validated pyrosequencing genotyping assays for APOE codon 112T>C and codon 158C>T polymorphic variants were used to genotype participant’s gDNA samples and identify APOE ɛ4 Carriers and APOE ɛ4 Non-Carriers status. By interrogating these two polymorphic variants, we identified the three APOE alleles: APOE ɛ2(TGC 112, TGC 158), APOE ɛ3 (TGC 112, CGC 158), and APOE ɛ4 (CGC 112, CGC 158). APOE genotype status has been determined for the enrolled cohort. A genome-wide-analysis study is underway and data expected to be available during the study. At ICL, whole blood is also collected in a PAXgene Blood RNA tube containing reagent for stabilisation of intracellular RNA, and stored −80°C. These samples will be used for future genetic analyses.
Medical history and clinical examinations (physical and neurological examination)
A thorough medical history was obtained including an evaluation of all body systems (ear, nose, throat (ENT), ophthalmic, musculoskeletal, gastrointestinal, urinary, respiratory, renal, cardiovascular, dermatological) with an emphasis on relevant medical history (eg, neurological, psychiatric, substance abuse, endocrine and metabolic). Safety and compatibility for neuroimaging were further ensured prior to the procedure.
Clinical examination included general physical examination and a separate comprehensive neurological physical examination. the general physical examination assessment included: general appearance, dermatological (including mucous membranes), ENT, cardiovascular, respiratory, abdomen, lymph nodes, musculoskeletal and any other findings. At neurological examination, mental status, cranial nerves, motor (strength), tone, involuntary movements, coordination (finger-nose, gait, postural reflexes and heel to shin), sensation (proprioception, cold, light touch), deep tendon reflexes, plantar reflexes and presence of other neurological signs (eg, tremor) were assessed.
Safety reporting
During the whole course of the study, new medical conditions and changes in medication were assessed at every site visit. All adverse events (serious and non-serious) were documented and reported according to the same protocol procedures applied in the main CHARIOT:PRO Main Study.11 Briefly, serious and non-serious events that occur from inception of participation all through to completion of last study-related procedure are captured and recorded for all participants. Events are judged as serious if fatal, immediately life-threatening; require hospitalisation or prolonging of existing hospitalisation; permanently (or significantly) disabling; a congenital anomaly or birth defect (in an offspring); or medically significant. Further data recorded for each suspected adverse event included the description (signs and symptoms or diagnosis), seriousness criteria, severity rating, duration (onset and resolution date), actions taken and outcome.
Study participant and public involvement
The ICL (via CHARIOT register) and EDI team have established Research volunteer panels consisting of lay members who met on an ad-hoc basis to support study development during the planning stage. These panels provided feedback on study design, procedures and dissemination for lay audiences. A newsletter is provided to study participants with updates regarding recruitment, study milestones and any important changes to the protocol. Participants were not directly involved in recruitment activities for the study.
CPSS participants further provide feedback on the experience of research participation at the different study visits, to ensure that their perspectives are represented in decision-making about the future of the project and to advise on planned study activities, including dissemination plans. Annual participant seminars are conducted for dissemination of study results and discussion of future plans. A newsletter is provided to study participants quarterly for study updates, as well as future plans. Participant input and feedback on volunteer experiences is typically encouraged for inclusion in the newsletter.
Ethical and regulatory considerations
To ensure the quality and integrity of research, CPSS is conducted in accordance with GCP Guidelines, GPPs issued by ISPE, applicable national guidelines, and to the Declaration of Helsinki 2013, as modified by the 52nd World Medical Assembly (WMA), Edinburgh, Scotland, 2000, and clarified by the WMA General Assembly, Washington 2002 and Tokyo 2004. The study has received approval from the National Research Ethics Service (NRES) Committee London Central (reference 15/LO/0711 (IRAS 140764)), as well as independent ethics review by committees from the local sites.
Informed consent
Formal informed consent is taken using an informed consent form (ICF) from both participant and study partners before participation in the study. Given the possibility that participants might lose mental capacity during the study; it was recommended at the outset of participation that the participant identified a Legally Authorised Representative (LAR). A LAR may include the spouse, a person specifically appointed to take care of the legal interests of the participant, an individual with guardianship, and a healthcare proxy, who provides consenting for research studies which is within the legal scope of the proxy’s delegated responsibilities (according to local applicable laws). The LAR must have the cognitive and mental capacities (as determined by the site investigator) enabling him/her to understand the procedures, risks and benefits involved with the study. The consent was given, and the form signed, at the initial visit or at follow-up visits at the study sites, based on the choice of the participant, and, where necessitated, on the choice of the LAR.
Duty of care
As part of the duty of care during the study, all clinically relevant information is shared with study participants where relevant and, with participant’s consent, communicated to the GP for medical follow-up. The clinically relevant findings shared included systemic hypertension and significant changes in cognitive assessments where the investigator felt they were relevant.
Confidentiality
Participant confidentiality is strictly maintained. Each participant is assigned a unique participant identifier on study enrolment, which is used for all subsequent data analysis and reporting. Participants’ National Health Service (NHS) numbers are collected and stored in keeping with industry standards for encryption/data protection, allowing for subsequent data collection from electronic health records in primary or secondary care within NHS. This data collection only occurs following NRES approval. All parties ensure that participant personal data is not included on any study forms, reports, publications, or in any other disclosures, except where required by law. The Investigators in compliance with Federal regulations, other applicable laws and International Conference on Harmonization (ICH) GCP Guidelines keep documents that are not for submission to the sponsor and/or its designee (eg, signed ICFs and Participant Information Sheets) in strict confidence. In accordance with regulations in the UK, participants are informed about data handling procedures.
Data management, analysis, and dissemination plans
Data management
The CPSS is conducted in accordance with Good Clinical Practice (GCP) Guidelines as such data are recorded and stored in a way that could be verified and reported in an accurate manner. All essential documents are filed in the trial master file/investigator site file. Source documents are kept in both paper and electronic formats. The main Electronic Data Capture system used in the current study is Medidata Rave. Both paper and electronic data are subject to daily and monthly internal audits based on standard operating procedures (SOPs). In addition, the investigator site files, paper source documentation and electronic source data are routinely monitored to maintain data accuracy collection to the highest degree.
Statistical analysis
Assuming the 3.5 year change from baseline in the G-PACC score has a SD of 2.4 for the Aβ positive participants,27 a sample size of n=250 with a 3.5-year dropout rate of 31% (ie, 10%/year) ensures the 95% CI for the 3.5-year mean change in G-PACC score in Aβ positive participants to be no wider than 0.72, assuming that the sample mean follows a Gaussian distribution. Analysis of change in G-PACC and RBANS over time will be performed with mixed models for repeated measures (MMRM) which assumes that missing data due to dropout are missing-at-random (MAR). The robustness of the analysis with respect to deviations from the MAR assumption will be evaluated. Analyses of the accruing results may be performed periodically while the study is ongoing. Analyses will generally be descriptive, but inferential analyses might be performed as needed. Potential unblinded interim analyses include:
Analyses for baseline characterisation of participants.
Analyses for determining longitudinal change in study endpoints once the last ongoing subject completes the month 12, month 24 and month 36 visits. These analyses will include descriptive statistics (n, mean, SD) and/or proportions for the Aβ positive and negative groups, but the Aβ status of individual participants will remain blinded.
Discussion
With the preclinical disease stages being increasingly recognised as the best timing for intervention, it is paramount that trial evaluations are sensitive enough to detect and track cognitive, functional and biological changes emerging in these stages while also possessing sufficient efficacy to detect therapeutic effects for drug trials. Furthermore, there is an urgency to identify robust and sensitive predictors of clinical progression in order to estimate individual risks for clinical AD and develop and apply therapeutic strategies prior to emergence of clinically evident AD dementia. Although an ambitious project, some limitations of this work are worth mentioning. The amyloid positivity rate is low and due to a need for an equal number of participants in each group (amyloid positive; amyloid negative), a high number of participants (78.6%) were excluded from the longitudinal follow-up phase. As a mitigating measure, enrichment criteria were introduced, with requirement of first-degree family history in volunteers aged 60–65 years old. The conduct of the study at only two sites is not typical of multisite international trials; on the other hand, this minimises several sources of variability that are independent of ageing and incipient Alzheimer’s disease (eg, inter-rater variability and differences in psychometric equivalence among different translations). It could be argued that the cognitive battery set may not be sensitive in predicting AD in healthy older adults, since these mostly tax modalities associated with AD dementia diagnostic criteria. Nonetheless, the high frequency (quarterly) follow-up of participants will facilitate determination of those assessments most sensitive for identifying the earliest signs and symptoms of AD-dementia and offers an opportunity to assess other performance parameters (eg, qualitative errors, lack of practice effect; speed-accuracy tradeoffs) that may indicate changes in cognitive and/or cerebral integrity in the lead up to AD dementia.28 Similarly, other assessments of physical health pertinent to AD risk, such as gait, hearing, or dental health are not included in our study. However, we do collect extensive medical history information at baseline and follow-ups that includes clinical abnormalities (eg, mobility issues; hearing impairment) that may be useful in our analyses.
The CPSS contributes towards this global agenda of AD-dementia prevention. The study features detailed and frequent clinical and cognitive assessments in a deeply phenotyped, presymptomatic cohort of older adults. An overall aim of the study is to prospectively compare changes in cognition, and other clinical measures, between individuals with presence of pathological levels of brain Aβ detected in PET scans or CSF and those without such evidence. CPSS also introduces a novel cognitive composite, the G-PACC, as a possible endpoint for future clinical trials. In this way, CPSS will expand on prior retrospective investigations of proposed cognitive composites,27 by prospectively investigating the longitudinal change of the components of the G-PACC composite. The performance of the G-PACC to detect effects will be compared against another cognitive composite and its subtests, the RBANS. The addition of the RBANS subtests, alongside other clinical data, will allow for the exploration of novel cognitive risk profiles for the progression of future AD. The baseline data will determine which measures are most sensitive for predicting longitudinal AD-related cognitive decline, informing future screening methods for clinical trials. The study includes both patient and proxy versions of functional interviews, such as the CFI and ADCS-ADL, to investigate longitudinal changes in everyday functioning in preclinical-AD individuals alongside cognitive decline and clinical characteristics. Dietary patterns and other lifestyle variables will also be assessed to consider the impact of environmental exposures on AD development. Therefore, the CPSS will also allow for the exploration of environmental and lifestyle predictors of cognitive decline and impairment.
The uniqueness of this study lies in its breadth and frequency (every 3 months) of assessments, as well as the planned explorations and comparisons of proposed cognitive composites for AD detection and tracking. The prolonged and detailed follow-up data offers opportunities for precise disease modelling and the evaluation of several methodological controversies within clinical trial design, such as the influence of practice effects on cognitive performance, in addition to mechanisms of reserve and resilience against cognitive senescence.
To date, CPSS has successfully completed its enrolment of 519 participants across two UK research sites, from 2451 screened volunteers. Next steps in CPSS’s milestones include the exploration of the baseline data for initial comparative analyses between stratified participant groups. The CPSS will continue as a multinational and multidisciplinary collaboration between industry, academia and the NHS to promote greater understanding of the aetiology of AD pathological attributes and symptom development, and champion the search for effective preventative therapies. Future plans include study extension to at least 4.5 years, at the ICL site, with addition of Tau-PET and follow-up structural MRI, and extensive state-of-art fluid biomarker discovery explorations at multiple timepoints.
Ethics statements
Acknowledgments
The authors wish to thank the participants who continue to volunteer their time and commitment towards the CPSS. We thank the staff at various sites who assist in the collection of biological, cognitive and other clinical data, and we thank the sponsor for funding this work. Imperial College London is grateful for support from the NIHR NW London Applied Research Collaboration.
References
Footnotes
Twitter @mecdoe
Contributors LM served as the ICL study site principal investigator. CWR served as the EDI study site principal investigator. CTU-M served as the lead author, with TW as co-lead. CTU-M, MK, JC, RP, GP, CAd-JL and HW were coinvestigators at School of Public Health, Imperial College London. KR was coinvestigator at Centre for Dementia Prevention, University of Edinburgh. All principal and coinvestigators contributed to the study design, coordination, data acquisition, development and critical review of the manuscript. DS and LB were employees at Bioclinica, and developed the imaging protocol for the study, and participated in development and critical review of the manuscript. Prof Majeed codeveloped the Chariot Register at School of Public Health, Imperial College London. DB, CR, MC, PG, DK, YCS, DP, HM-Y, KL, JC and CL were study investigators at School of Public Health, Imperial College London; TW and NR-M were study investigators at Centre for Dementia Prevention, University of Edinburgh, and all contributed to data acquisition and review of the manuscript. GN, MR, MA, NK, NR, ZS, RB and MF contributed to the study conception, protocol design and development. GN, Sb, Kacher, ZS and MF contributed to study design and were responsible for data review, interpretation, and development of the manuscript. In addition, MA was project pharmacoepidemiologist and NR, JD and Kacher served as project biostatisticians, responsible for aspects of study design, statistical data analysis and statistical input. ZS served as the unblinded statisticaian and imaging lead for the project. All authors provided direction and comments on the manuscript, made the final decision about where to publish this protocol and approved the final draft and submission to this journal.
Funding This work was supported by Janssen Pharmaceuticals Research & Development, LLC.
Competing interests MTR was formerly employee of Janssen and is an industry consultant. GN, SB, HMA, MF, KK, ZS and NR are employees of Janssen Research & Development, LLC and own stock/stock options in the company. NK and HRB are former employees of Janssen Research & Development, LLC and both own stock/stock options in the company. JD is an employee of Janssen China Research and Development Center and owns stock/stock options in the company. LM served as principal study investigator at Imperial College of London (ICL) abd has held consultancy agreements, in the last five years, with Eli Lilly, Astra Zeneca and Takeda and is National Coordinator for the TOMMORROW, Amaranth and Generation Clinical Studies; and does not hold any agreement with any of the funders in relation to patents, products in development relevant to this study or marketed products. CR served as principal study investigator at Edinburgh University (EDI), and has served as a consultant in the last five years for: Actinogen, Biogen, Roche Pharmaceuticals and Roche Diagnostics, Eisai, Abbott Pharmaceuticals, Eli Lilly, Kyowa Kirin, Signant Health, Merck and Nutricia. He is also the Chief Investigator and Co-coordinator of IMI-EPAD which is a public-private partnership of 39 partners www.ep-ad.org. CU-M, JC, RP, GP, Cd-JL, HW and MK served as co-principal study investigators at ICL for Janssen Research & Development, LLC and all declare no conflict of interest. JC's and AM’s posts at ICL are in part supported by the NIHR NW London Applied Research Collaboration. CR, DB, MC, PG, DK, YC-S, DP, LC, KL, HM-Y, JC and AM were study investigators at ICL and declare no conflict of interest. KR served as co-principal study investigator at EDI for Janssen Research & Development, LLC and declares no conflict of interest. TW and NR-M were study investigators at EDI and declare no conflict of interest. DS and LB are employees of Bioclinica Inc. and declare no competing interests.
Patient and public involvement Patients and/or the public were involved in the design, or conduct, or reporting, or dissemination plans of this research. Refer to the Methods section for further details.
Provenance and peer review Not commissioned; externally peer reviewed.