Article Text

Abstract
Objective To determine the feasibility and optimal design of a randomised controlled trial (RCT) of Seizure First Aid Training For Epilepsy (SAFE).
Design Pilot RCT with embedded microcosting.
Setting Three English hospital emergency departments (EDs).
Participants Patients aged ≥16 with established epilepsy reporting ≥2 ED visits in the prior 12 months and their significant others (SOs).
Interventions Patients (and their SOs) were randomly allocated (1:1) to SAFE plus treatment-as-usual (TAU) or TAU alone. SAFE is a 4-hour group course.
Main outcome measures Two criteria evaluated a definitive RCT’s feasibility: (1) ≥20% of eligible patients needed to be consented into the pilot trial; (2) routine data on use of ED over the 12 months postrandomisation needed securing for ≥75%. Other measures included eligibility, ease of obtaining routine data, availability of self-report ED data and comparability, SAFE’s effect and intervention cost.
Results Of ED attendees with a suspected seizure, 424 (10.6%) patients were eligible; 53 (12.5%) patients and 38 SOs consented. Fifty-one patients (and 37 SOs) were randomised. Routine data on ED use at 12 months were secured for 94.1% patients. Self-report ED data were available for 66.7% patients. Patients reported more visits compared with routine data. Most (76.9%) patients randomised to SAFE received it and no related serious adverse events occurred. ED use at 12 months was lower in the SAFE+TAU arm compared with TAU alone, but not significantly (rate ratio=0.62, 95% CI 0.33 to 1.17). A definitive trial would need ~674 patient participants and ~39 recruitment sites. Obtaining routine data was challenging, taking ~8.5 months.
Conclusions In satisfying only one predetermined ‘stop/go’ criterion, a definitive RCT is not feasible. The low consent rate in the pilot trial raises concerns about a definitive trial’s finding’s external validity and means it would be expensive to conduct. Research is required into how to optimise recruitment from the target population.
Trial registration number ISRCTN13871327
- epilepsy
- accident & emergency medicine
- organisation of health services
- health economics
- clinical trials
This is an open access article distributed in accordance with the Creative Commons Attribution 4.0 Unported (CC BY 4.0) license, which permits others to copy, redistribute, remix, transform and build upon this work for any purpose, provided the original work is properly cited, a link to the licence is given, and indication of whether changes were made. See: https://creativecommons.org/licenses/by/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
Randomisation was done remotely by computer and stratification factors and allocations concealed from those collecting baseline and follow-up data and analysing it.
Participants were recruited from site’s serving areas, where social deprivation was high and epilepsy control poor and so similar to those where a definitive randomised controlled trial (RCT) would likely need to focus recruitment.
We completed one of the few microcostings of a self-management intervention for epilepsy.
Despite recruited patient participants stating sufficient emergency department use when screened, routine data subsequently suggested ~40% did not meet the inclusion criteria.
We estimated the effect of Seizure First Aid Training For Epilepsy only on the proposed primary outcome measure for a definitive RCT.
Background
International evidence shows a significant minority of people with epilepsy (PWE) frequently use hospital emergency departments (EDs).1–4In the United Kingdom, around 20% of PWE visit each year, of which half are admitted.5–7 While the exact distribution of use among attendees is unclear,8–10 ~60% may make several visits each year.11
Emergency hospital use by PWE has been identified as an area for potential cost savings,12as while expensive—approximately ~£70–90 million in England each year8 13 14—it is often of little clinical benefit since most attendees have known, rather than new epilepsy and experienced uncomplicated seizures.9 15 16 Guidance states such seizures could be managed by PWE and their significant others (SOs).17 18 Indeed, iatrogenic harms may arise in seeking emergency care for them.
PWE visiting EDs have a characteristic profile. They report more seizures, anxiety, poorer quality of life and are more likely to live in socially deprived areas compared to the wider epilepsy population.11 19–22 They therefore share some of the characteristics of those at increased risk of epilepsy-associated death.23 24 In the UK, PWE visiting EDs can also be challenging to identify for research since most (~62%) are not being followed up by an epilepsy specialist,1 primary care providers are also not always notified of ED visits by their patients9 and because EDs do not always code the reason for an attendance in sufficient detail.25
While the focus on PWE attending EDs is welcomed, it needs to be translated into care improvements. Although a range of promising interventions have been suggested,26 assessment of their utility is lacking.27
One intervention proposed is seizure first aid training. It has potential as PWE frequently visiting EDs and their SOs (to whom care decisions can be delegated28 29) express particularly low knowledge and confidence in this domain, are fearful of seizures and there are indications this leads them to seek emergency medical attention for seizures, rather than self-managing them.30
As no such intervention existed, we developed one—called Seizure First Aid Training For Epilepsy (SAFE). SAFE is a manualised group-based self-management course (table 1). It’s rationale and development has been reported.31 In brief, it was codesigned with health professionals, patients and carers. It was developed for delivery to groups of up to 10 patient–carer dyads by a single facilitator with knowledge of epilepsy and lasts for ~4 hours. It contains six modules centred around basic epilepsy and first aid knowledge, the recovery position, informing others about epilepsy and how to help if seizures occur, medical identification, seizure triggers and home safety. Materials include presentation slides and professionally produced videos. Its behaviour change potential has been optimised by course recipients completing a self-affirmation exercise at the start.
SAFE course and its content
SAFE’s efficacy now needs testing. A randomised controlled trial (RCT) would be an appropriate methodology for this. ED use over the 12 months following randomisation could be the primary outcome. It is unknown whether such a trial is feasible and what its optimal design should be.
Specifically, will patients and their SOs take part, attend SAFE and be willing and able to be followed up? It is also not possible to calculate the required sample size because the distribution of ED use is unclear. These uncertainties exist because SAFE is newly developed and no RCT has attempted to recruit the target population.
It is also unclear how to measure ED visits. Funders are encouraging trialists to use routinely recorded data to assess outcomes. In England, routine data on a person’s ED use are recorded within the Hospital Episode Statistics system. This data’s cost, how long it takes to obtain and its comparability to self-report are unknown.
In such circumstances, guidelines highlight the importance of pilots.32 Their primary focus is not effect, but judging feasibility and providing information to optimise a definitive RCT’s design.33 We thus completed a pilot RCT comparing SAFE plus treatment-as-usual (TAU) or TAU alone. It had the following objectives, which will be relevant to those interested in SAFE and those researching this population: (1) estimate likely eligibility, consent and retention rates in a definitive RCT; (2) estimate annual rate of ED visits in TAU group and the likely dispersion parameter; (3) determine feasibility of measuring ED use by routine data; (4) estimate completion rates of study assessment tools; (5) estimate rates of researcher unblinding; (6) provide summary statistics to estimate effect of SAFE on ED use and its precision; (7) capture patient’s and SO’s views on trial participation; and (8) estimate the intervention’s cost.
Methods
The trial’s protocol has been published.30 Here we provide a brief overview.
Design
Parallel arm, multi-centre, external pilot RCT. Assessments with participants on the definitive trial’s proposed primary and secondary outcome measures were performed at baseline prior to randomisation (T0) and 12 months later (T3, table 2). Interim assessments occurred at 3 (T1) and 6 months (T2).
Proposed primary and secondary outcome measures for a definitive trial used within pilot trial by assessment and participant type*
Supplemental material
SAFE was offered to the TAU alone group after T3 assessments were completed.
Trial setting
Three hospitals in north-west England—which serve populations featuring high-social deprivation34 35 and emergency admissions for epilepsy21—were recruitment sites (see Acknowledgements section). From May to December 2016 patients were invited who had attended any of these hospital’s ED over the prior 12 (and with governance approval, later extended to 18) months for epilepsy (see Patient recruitment section).
Ethical considerations and approvals
The National Research Ethics Service Committee (15/NW/0225) and Health Research Authority (166241) approved the study. An independent Trial Steering Committee monitored the trial.
Patient recruitment
ED clinicians searched their hospitals’ attendance record systems for potential participants (online supplementary file 1), screened their triage cards and posted invitations to eligible patients. Recipients had 3 weeks to return an opt-out response if not interested in participating. Those not opting-out were telephoned by a researcher to verify eligibility and willingness to participate. Patients taking part (and their SO if they chose to take part with one) provided informed consent at an enrolment appointment with a researcher (DS). For patients, this included consent to access their routine data.
Eligibility criteria
Table 3 details the criteria. In brief, patients were eligible if they were aged ≥16, diagnosed with epilepsy, prescribed antiepileptic drugs, could give informed consent and, when telephoned, self-reported ≥2 ED visits for epilepsy in the prior 12 months.
Participant inclusion and exclusion criteria
Randomizsation and blinding
Patients (and their SOs) were randomised (1:1) by an online system managed by the Liverpool Clinical Trials Centre. It used a minimisation programme with a built-in random element and two stratification factors (recruitment site and whether the patient reported epilepsy stigma at baseline).
Usual care provider(s) and DS, who was responsible for recruitment and follow-up, were blinded to allocations and stratification factors. Participants could not be blinded and so were asked (and reminded at follow-ups) not to reveal their allocation to DS.
Staff (GM) organising attendance at SAFE were not involved in data collection and not blinded. The trial statistician (SN) and senior statistician (CTS) were blinded until the database was ‘locked’.
Measures to assess patient and SO participants’ outcomes
Table 2 details these measures.
Intervention
An epilepsy nurse specialist (JB) delivered SAFE within a local hospital’s educational centre. A fidelity assessment found they delivered SAFE with excellent protocol adherence and competence.36
Outcomes
To achieve the study objectives, rates of eligibility, consent and retention were calculated. Retention being the percentage of patients for which 12-month primary outcome data were secured. Two a priori progression criteria helped judge feasibility: (1) ≥20% of eligible patients were recruited; (2) 12-month primary outcome data were secured for ≥75% of patients.
By assessing participants on the proposed outcome measures, we obtained estimates of the distribution of ED use, measure completeness and SAFE’s effect. To evaluate blinding, DS completed a ‘Treatment Guess’ form after each participant’s T3 assessment or withdrawal. It required her to state which treatment arm she believed the participant had been randomised to. The proportion of participants willing to participate in such a trial again was ascertained and experience of adverse events calculated. Time taken to obtain routine data and the fee payable were recorded. To see if self-reported ED visits provided a reliable estimate compared with routine data, the agreement between the two data sources on how many ED visits a patient had made was explored.
Statistical analyses
As this was a pilot RCT, a power calculation was inappropriate. Instead, the sample size was chosen to provide adequate precision to estimate the parameters required.37
Based on existing data,1 14 16 38 39 it was anticipated that 12 months of attendances from each ED would allow identification of ~400 eligible patients. With a 20% consent rate, 80 patient participant accruals could be expected. With 80 patient participants we could estimate a potential drop-out rate of 25% to within a 95% CI of ±10% and a consent rate of 20% to within a 95% CI of ±4%. Assuming ED data at T3 was not available for 25% of patients, data from 60 patients would still allow robust estimation of the ED rate and dispersion parameter. Sample sizes of 24 to 50 have been recommended as ‘adequate’ for pilot studies.37 40
Analyses were documented a priori in an analysis plan and performed using SAS (V.9.4). Baseline characteristics for patient and SO participants are described using descriptive statistics and patient participants compared, on a subset, to eligible patients declining participation. Parameters are reported with 95% CIs.
SAFE’s effect on ED use, with and without adjustment for number of ED visits prior to randomisation, was estimated using negative binomial regression (NBR) on a modified intention-to-treat basis (as defined by Del Re et al.41). Participants were included with their number of ED visits recorded with no data imputation. NBR was the prespecified statistical approach as over-dispersion (ie, large variance) was anticipated in the number of ED visits reported. Between-group differences are presented as rate ratios and, as per recommendations for hypothesis testing within pilot trials,42 tested according to 5, 10% and 20% levels of significance.
The proportion of correct treatment guesses was determined and Cohen’s Kappa computed. Bland-Altman plots compared ED visits as measured by routine data and self-report.43
As no consensus exists about what constitutes a clinically important reduction in ED visits,44 average annual rate of ED visits in the SAFE+TAU and TAU alone groups postrandomisation (measured according to routine data) and the likely dispersion parameter from the adjusted NBR model were used to estimate the sample size of a definitive trial using Keene et al’s45 formula. According to the formula, the number of patient participants required per group in a definitive trial to detect the size of the effect shown in the pilot study is:

where Z1−α/2 and Z1−β are critical values of the normal distribution for specific values of alpha (α) and power (β). µ1 and µ2 are the estimated ED rates from the two treatment groups and k the negative binomial shape parameter from the associated gamma distribution which explicitly represents variability between subjects. For the calculations, alpha was set at 5%, but the dispersion parameter and power required was varied to explore differences in sample size required.
Microcosting
Microcosting adopted the perspective of an academic non-profit making institution and was conducted in three steps46: (1) resource identification; (2) resource use measurement, applying the time and motion method47; and (3) valuation using price year 2017/2018 for local and national data. Data were analysed using Microsoft Excel 2010 to calculate the fixed and variable costs of delivering SAFE. Fixed costs included, equipment, website, freepost licence, venue hire, facilitator staff cost and facilitation resources; and assumed 11 groups/year and equipment life of 1 year. Variable costs were support staff and office costs, staff and participant travel expenses and consumables. Total cost per group was calculated as fixed costs plus variable costs, for each group and each arm (SAFE+TAU; TAU). Mean cost per group was calculated as the sum of total costs/number of groups. Mean cost per delegate (or patient only) was calculated as sum of total cost per group in each arm/sum of delegates (or patients only) in each arm. Results are presented as cost per training session, mean cost per delegate and mean cost per patient. The 95% central range (CR) for costs and differences were generated using Monte Carlo Simulation of 10 000 replications.
Patient and public involvement statement
This research came about as improving education for patients and families on epilepsy is a top research priority.48 To ensure SAFE was developed and tested in a way that met service users' needs the ‘Epilepsy Society’ and patient and SO representatives from the ‘Brain and Spine Foundation’ helped develop recruitment materials; the pilot was overseen by a Trial Steering Committee including two service user representatives; SAFE was designed by an Intervention Panel including two service user representatives and its content informed by feedback from 23 service users31; finally, pilot trial participants reported on the burden of participation with a view to optimising the design of a potential definitive RCT.
Results
Participant recruitment, allocation and treatment
Of the 4016 individuals identified, 1220 (30.4%, 95% CI 29.0% to 31.8%) had visited for established epilepsy. Of these, 424 (34.8%, 95% CI 32.1% to 37.4%) were eligible; eligibility rate 10.6% (95% CI 9.6% to 11.5%, figure 1, online supplementary file 2).

CONSORT diagram of flow through the pilot trial. CONSORT, Consolidated Standards of Reporting Trials; SOs, significant others.
Of the 424 patients invited, 53 consented; consent rate was 12.5% (95% CI 9.3% to 15.6%). Telephone contact could only be made with 203 (47.8%, 95% CI 43.1% to 52.6%) patients. The consent rate among those who could be contacted was 26.1% (95% CI 20.0% to 32.2%). The main reasons for 150 patients declining participation were ‘lack of interest’ (42.7%, 95% CI 34.8% to 50.6%) and being ‘too busy’ (22.6%, 95% CI 16.0% to 29.4%).
Of the 53 consenting patients, 51 were randomised (with 37 SOs, figure 1 gives reasons for non-randomisation). Of the 51, 26 (and 18 SOs) were allocated to SAFE+TAU and 25 (and 19 SOs) to TAU alone. Most (20, 76.9%) patients and SOs (13, 72.2%) randomised to SAFE+TAU attended a course.
Participant demographics and epilepsy characteristics
The patient participants mean age was 39.9 years (SD 15.7, range 16–71); 29 (56.9%) were female and most (74.4%) lived in areas high in deprivation (49% in the 10% most socially deprived areas in England, table 4).
Characteristics of eligible patients who did and did not agree to participate
Recruited patients were comparable in age and deprivation to those declining participation. Females were over-represented (table 4).
Recruited patients had an epilepsy diagnosis for a median of 17.3 years. Most (62.8%) patients reported ≥10 seizures in the previous year and having seen a neurologist (74.5%, table 4). Participants’ mean Quality of Life-31-P score was low at 48.3 (SD 17.3; range 17.1 to 79.5). Twenty-six (50.9%) patients had ‘probable’ clinical anxiety. Most (n=42, 82.3%) patients reported feeling stigmatised by epilepsy; 15 (29.4%) highly. The treatment groups were broadly similar in demographics and epilepsy characteristics.
SO participants were typically a partner/spouse (43.2%) and most (89.2%) had daily patient contact (online supplementary file 3). With a mean Zarit Caregiver burden score of 18.9 (SD=12.51), SOs typically reported ‘little or no burden’49 but anxiety was high50; 15 (40.5%) SOs had ‘probable’ clinical anxiety.
Participant retention
Proposed primary outcome measure
Of the randomised patients, 3 (5.8%) formally withdrew over follow-up, removing access to their routine data. Primary outcome data on ED use at 12 months (and for the 12 months prior to randomisation) were available for the remaining 48 patients, giving a retention rate of 94.1%.
Proposed secondary outcome measures
Thirty-seven (72.5%) randomised patients and 21 (56.8%) SOs attended their 12 months questionnaire appointment (T3). The extent to which measures were completed at these and the interim appointments varied (online supplementary file 4). Self-report data on ED use at T3 was obtained from only 34 patients, giving a retention rate on this measure of 66.7% patients.
ED use
Baseline, prior to randomisation
Routine data for the 48 patients for whom consent was maintained showed they made 122 ED visits in the 12 months before randomisation (online supplementary file 5). The mean was 2.5 and median 2 (range 0 to 12). ED use was slightly higher for TAU participants (table 5).
Number of ED visits patient participants made according to routine data
Despite only consenting patients who when telephoned reported ≥2 ED visits in the prior 12 months, routine data indicated 4 (8.3%) had not made any visits during the prior 12 months. A further 19 (39.6%) made only one.
At 12 months, effect of SAFE
Compared with the 12 months prior to randomisation, mean ED use over follow-up reduced for the SAFE+TAU group from 2.1 visits to 1.8 (difference −0.3). For the TAU group, it increased from 3 visits to 3.4 (difference 0.4, table 5). Unadjusted NBR estimated the visit rate was 46% lower in the SAFE+TAU group compared with the TAU group (rate ratio=0.54; Vuong’s test z=−0.17, p=0.87). In the adjusted model, SAFE’s effect reduced (rate ratio=0.62). Between-group differences were not significant at the 5% or 10% alpha level in either the unadjusted or adjusted model. The effect was significant in both at the 20% level (table 6). The dispersion parameter under the adjusted NBR model was k=0.69 (CI 0.17 to 1.21).
Between-group intervention differences in number of ED visits
Obtaining routine data and its correspondence with self-report
Routine data took 8.5 months to secure, arriving ~9 months after T3 assessments finished. The direct cost was £6960. Online supplementary file 6 shows substantial work on behalf of the research team to obtain the data was required. In one instance, an appeal against a decision to reject the application—5 months in—was necessary.
Routine data did not match patient self-report in most cases. Forty-two patients had self-report and routine data on ED use at baseline. Only 3 (7.1%) reported the same number of visits as their routine data indicated. Most (76.2%) patients reported more (by 3.8 visits on average, figure 2A). There was greater agreement between routine and self-report data at T3; 11 (32.4%) patients reported the same number of visits as their routine data (figure 2B).
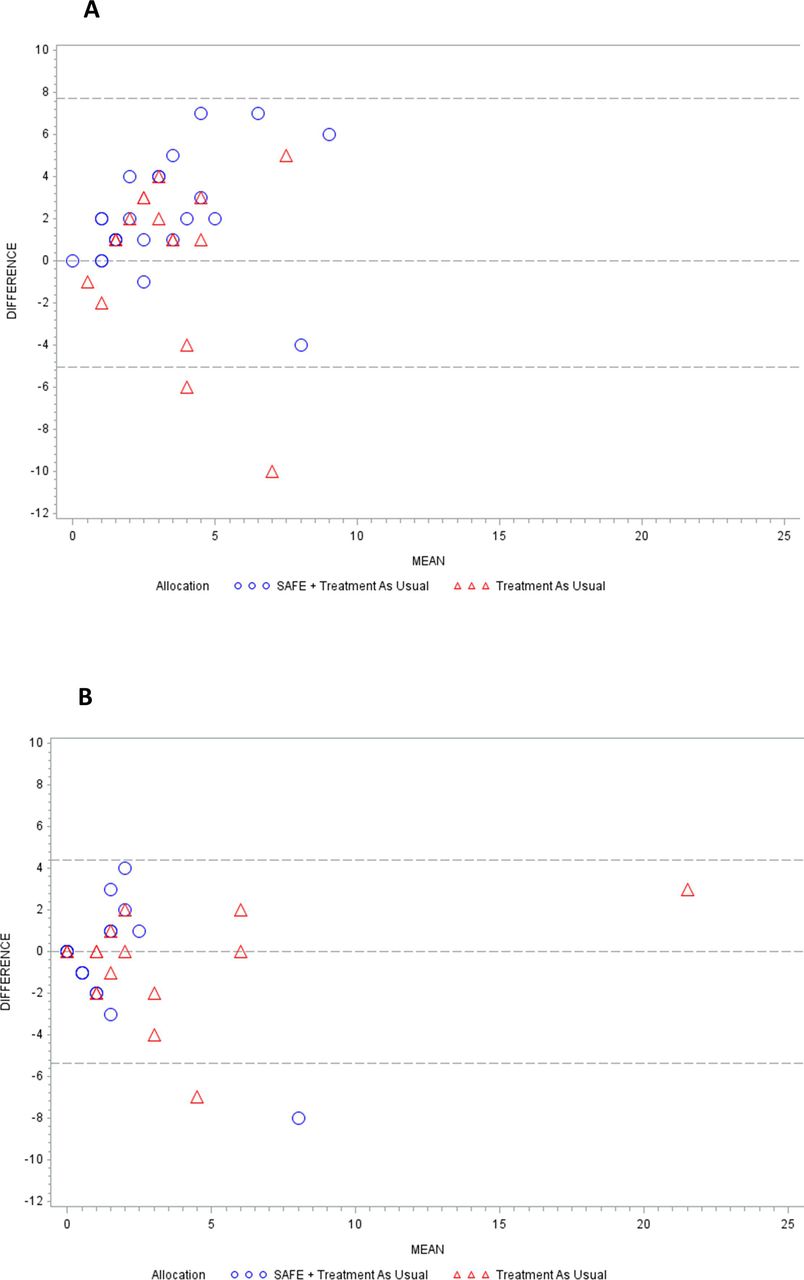
{kind=link}
{kind=link}
(A) Bland-Altman plot of agreement between self-reported ED use and routine data on ED use in 12 months prior to randomisation. (B) Bland-Altman plot of agreement between self-reported ED visits and routine data on ED use over 12 months following randomisation. ED, emergency department; SAFE, Seizure First Aid Training For Epilepsy.
Blinding
The researcher correctly guessed which of the two treatment arms 35 patient participants had been allocated to by the randomisation process; unblinding rate 68.6% (CI 54.1% to 80.9%). The chance-corrected kappa statistic of 0.37 (CI 0.12 to 0.63) equated to ‘fair’ agreement.
Safety
No serious adverse events related to participation occurred (online supplementary file 7).
Participant feedback
Thirty-two (68.1%) patients and 20 (62.5%) SOs completed the T3 feedback questionnaire. All but one said they would participate in such a trial again; participants indicated they perceived benefits from SAFE.
Sample size calculation
Based on the estimated effect of SAFE (see At 12 months, effect of SAFE section), table 7 shows the number of patient participants required per group for a definitive trial. If the central value in the estimate range for the dispersion parameter k of 0.69 is used and 90% power stipulated, then a total starting sample of 674 patient participants (ie, (308×2)+58) would need to be recruited. This accounts for the 9.4% of recruited patients who (on the basis of the pilot trial) would be anticipated to withdraw consent to access their routine ED data. In the pilot trial, 5 of the 53 patients recruited withdrew consent.
Required sample size for a definitive trial to detect estimated effect of SAFE+TAU on ED use
Microcosting
Delivering SAFE was estimated to cost £333 (CR £288 to £380) per patient (with or without a SO). When including the cost SAFE’s development (£181 per person), the mean cost per attendee, based on all 55 participants in SAFE+TAU or TAU groups who ultimately attended a SAFE session, was £375 (CR £348 to £402, online supplementary file 8). This reduced to £194 (CR £167 to £221) when excluding sunk costs. Staff time accounted for 50.01% of the cost of SAFE’s delivery. SAFE’s facilitator was not local to the trial area. Thus, an analysis of facilitator costs without travel expenses (time and cost), reduced the mean cost by 21.08% from £194 (CR £167 to £221) to £152 (CR £124 to £179) per delegate (£261 per patient with or without SO). The annual fixed cost of setting up and running SAFE was £1122 (+£35.07 per patient, based on 32 patients/year attending with or without SO).
Discussion
The pilot trial was successful, providing estimates of key parameters, including recruitment and retention. An informed decision regarding feasibility and optimal design of a definitive trial of SAFE can now be made.
Positives from pilot for feasibility and design of definitive trial
Identification, treatment and safety
The pilot trial indicates it is possible to identify, consent, randomise and safely treat persons from the target population within a definitive trial. This was not a given since no RCT had focused recruitment on them.
Retention and measuring ED use
By using routine data as the basis of the primary outcome measure, a definitive trial should not be affected by attrition. It permitted 12-month data to be secured for 94% of pilot participants. Despite having shorter follow-ups (6 months), 151 definitive RCTs funded by the National Institute for Health Research () secured outcome data for 89% of patients.51 The use of routine data in our pilot trial meant it satisfied the retention progression criterion. It would not have been met if self-report data were relied on (only 67% of patients provided it). This leads us to recommend ED use be measured using routine data.
Another reason for the recommendation is its cost would be low compared with employing staff to obtain self-report data. Routine data would also prevent a definitive trial from exposure to apparent recall bias, with patients seemingly over reporting ED use (it has been asserted that bias is not an issue for ‘‘big ticket’’ service items).52 53
However, our pilot trial does caution that provision of routine data is slow and not straightforward. It took 8.5 months to obtain. In principle, such applications should be processed within 2 months.54 Data providers should attend to such issues as those funding and designing trials need confidence that data can be secured and realistic estimates. It is also worth noting that some of the ED visits attributed to our participants may not have been epilepsy related. Since a diagnostic code is not recorded for ~35% of ED visits, we asked NHS Digital to inform us of any ED visits recorded for our participants.25
Negatives from pilot trial for feasibility and design of definitive trial
Consent
Only a small proportion (10.6%) of identified patients were eligible and willing (12.5%) to take part, meaning the progression criterion that ≥20% of eligible patients agree to participate was not met.
The progression criterion was stated a priori because when uptake is so low there is the real possibility that those who are and are not taking part differ. We compared the age and deprivation of patients who did and did not take part and found no obvious differences. We do not know whether they differed on other indices since access to non-participants’ medical records was not ethically permissible. One indication the pilot trial might not have recruited a representative sample was the high proportion of patients seeing a neurologist in the prior 12 months (~75%). Evidence suggests this should be closer to 38%.1
The consent rate also raises questions over the likelihood of a definitive trial being funded since it would make it expensive. On average, each pilot site generated 17.6 patient accruals from 18 months of attendances. To achieve a sample size of ~674 PWE, a definitive trial could thus require ~39 sites (half of England’s EDs). This is 2.5 times more sites than in recent NIHR funded RCTs52 which already had a mean cost of ~£1.3 million.55 A definitive RCT of SAFE might thus not represent acceptable value for money to funders.56
Epilepsy and its consequences (eg, seizures, memory difficulties, no driving license) make recruiting PWE challenging. The consent rate in the pilot is nevertheless low. In the largest RCT of self-management (Self-Management education for adults with poorly controlled epILEpsy [SMILE] trial), 37% of the people with uncontrolled epilepsy invited took part.57 The characteristics of the patients from ED agreeing to participate in our pilot suggest that one reason for the low uptake might be stigma; 82% felt some (21.6% felt highly stigmatised). This is higher than in the wider epilepsy population; 63% of SMILE’s sample felt stigmatised (12.5% highly).58 Stigma can make PWE feel ashamed and reluctant to talk about epilepsy. This could explain why the target population is so challenging to recruit. Unfortunately, it is unclear how to revise recruitment to mitigate against this.
Evidence-based strategies were employed in the pilot to maximise recruitment,30 and invitation materials coproduced with patients. It is generally considered preferable for a usual care provider, with whom a patient has an established relationship, to invite a person into research. Difficulties identifying the target population (see Background section) meant we had local ED clinicians do the inviting. A future trial might consider asking EDs to identify ostensibly eligible patients, but for the general practitioners of the identified patients to do the inviting. This may boost recruitment.59
Effect of intervention
Our pilot trial estimated SAFE’s effect to be modest (reducing ED visits from 2.1 over 12 months to 1.8). This has negative consequences for a definitive trial, not least because it requires it to have a large sample to detect the effect. Efficacy was not our pilot trial’s primary focus and the estimate may be imprecise. Indeed, it might be that those who declined to participate in the trial and who appeared to differ in some important ways, might have benefited more. Previous evidence does suggest it is in the region expected.
Specifically, only one RCT—by Sajatovic et al60—has considered change in ED use following epilepsy self-management.61 Conducted in the USA, it compared ‘Self-MAnagement for people with epilepsy and a histoRy of negative health evenTs' (SMART)—an 8-session group intervention—to wait list control. No significant change between groups was found in subsequent ED/hospitalisation use. For SMART, it reduced from a mean of 1.22 by 0 .44 over the 6 months following randomisation. For the controls it reduced from 2.4 by −1.26 visits.
Judgement regarding progression to definitive trial
Thabane et al62 provide a framework for judging whether to progress to a definitive trial. In satisfying only one progression criteria, a definitive trial based on the pilot trial’s design is not feasible. We have also not identified any modifications that will make it feasible. We therefore recommend not proceeding.
Strengths and limitationss
Strengths include the pilot trial being reported according to guidelines,63 allocations being concealed, that patients were recruited from sites similar to those likely for a definitive trial and we included a microcosting of SAFE’s delivery.
The pilot trial is not without potential weaknesses. Most importantly, despite recruited patient participants stating sufficient ED use when screened, routine data subsequently suggested ~40% did not meet the inclusion criteria. This could limit our findings’ external validity and attenuate SAFE’s effect. Another potential limitation is we estimated the effect of SAFE only on the proposed primary measure. We did not estimate its wider effects, including on duration of ED visits and admissions.
Conclusion
A definitive trial of SAFE is not currently feasible. Research is required to determine how people from the target population can be better recruited.
Acknowledgments
We thank the people who kindly participated in this study and the local ED sites for their support—namely, Arrowe Park Hospital (Wirral University Teaching Hospital NHS Foundation Trust) and Aintree University Hospital and the Royal Liverpool University Hospitals (Liverpool University Hospitals NHS Foundation Trust). Other persons who contributed to this work are Mrs Juliet Bransgrove (JB, epilepsy nurse specialist), Dr Duncan Appelbe (website development/management) and Ms Gail Moors (GM, Administrator). We also acknowledge the support we received from our Steering Committee (Professor Alasdair Grey (chair), Professor Pete Bower, Dr Paul Cooper, Mr Mike Jackson, Ms Helen Coyle, Mr Mike Perry, Mrs Linda Perry, Mrs Jayne Burton and Mr Sam Burton). The study sponsor was the University of Liverpool (reference: UoL001108) and the Clinical Trials Research Centre’s registration number is 12.
References
Footnotes
Twitter @sjn_16
Contributors AJN and LR conceived the study and designed it together with AGM, CTS, DAH, MM and SG. SN and CTS planned and conducted the statistical analysis, EAH and DAH led with the economic analysis. AJN was the chief investigator, with MB, JM and EM leading participant identification and DS recruitment and follow-up. AJN wrote the manuscript, with revisions and approval of the final manuscript coming from all authors.
Funding This project was funded by the National Institute for Health Research’s Health Services and Delivery Research Programme (HS&DR Programme, project number 14/19/09). The views and opinions expressed herein are those of the authors and do not necessarily reflect those of the University of Liverpool, the HS&DR programme, the NIHR, the NHS or the Department of Health and Social Care.
Competing interests None declared.
Patient and public involvement Patients and/or the public were involved in the design, or conduct, or reporting, or dissemination plans of this research. Refer to the Methods section for further details.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request. Requests for anonymised data should be submitted to the corresponding author.